Non-Steroidal Anti-Inflammatory Drugs in Alzheimer's Disease and Parkinson's Disease: Reconsidering the Role of Neuroinflammation

Abstract
:1. Introduction
2. NSAID Mechanism of Action



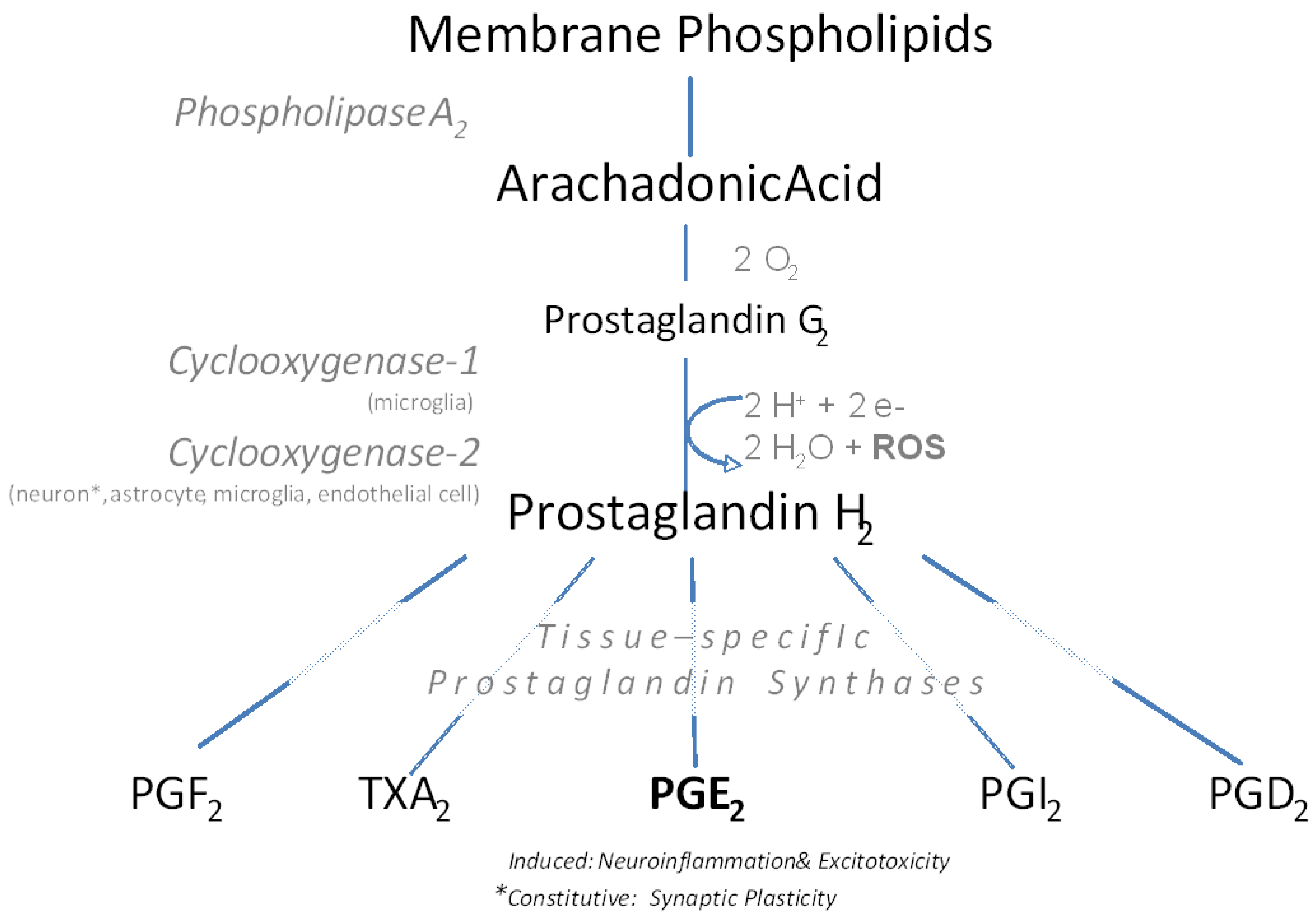{kind=link}
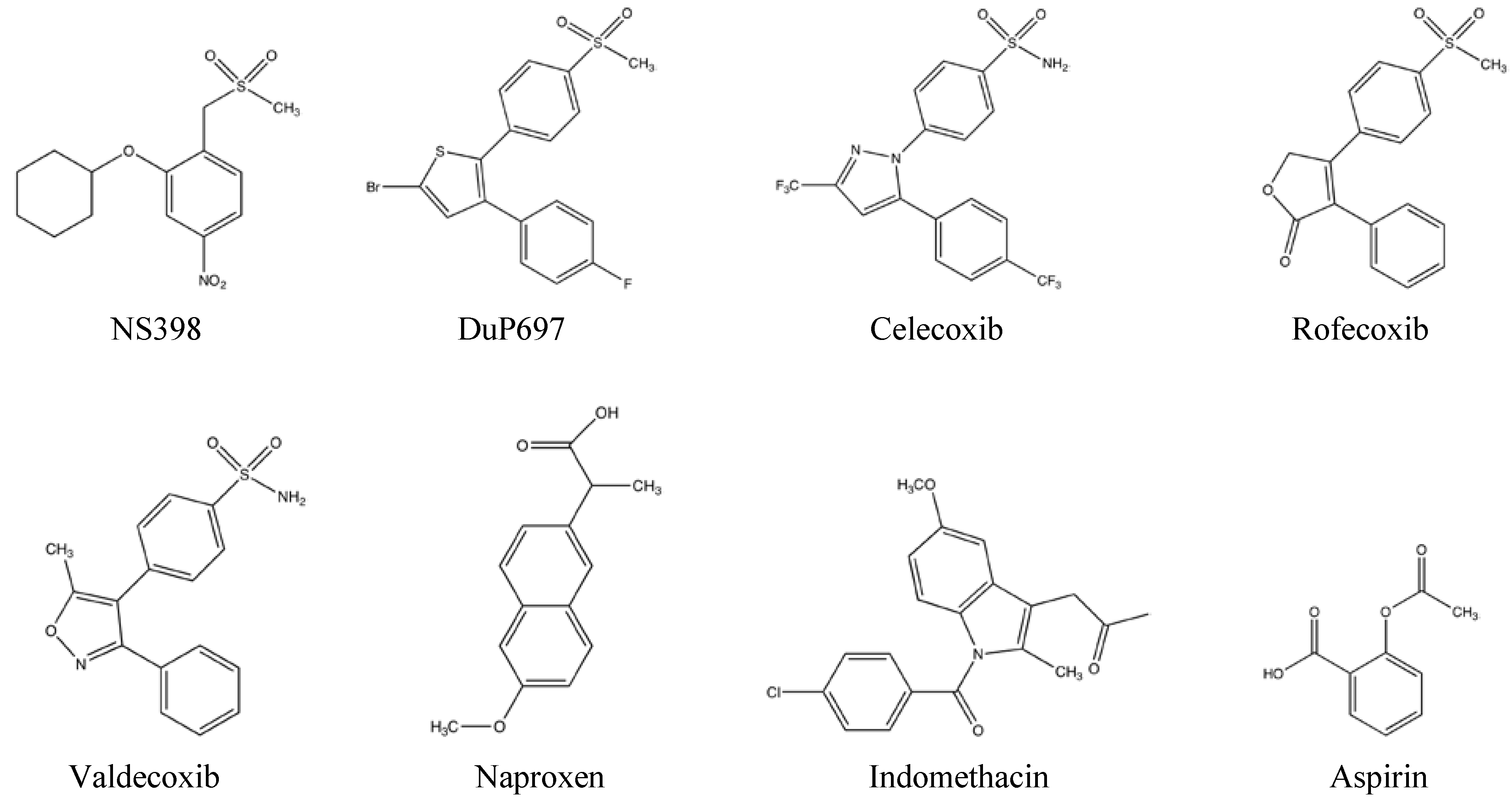{kind=link}
3. Overview of Neuroinflammation
4. Alzheimer's Disease
4.1. Disease impact and pathology
4.2. Animal models investigating neuroinflammation in AD
4.3. Clinical trials of NSAIDs in AD treatment and prevention
4.4. Consideration of clinical trials to determine future investigation of NSAIDs in AD
| Author | Year | Ref. | Patient description | Total number of patients recruited | Total number of patients included in analysis | NSAID | NSAID duration (months) | NSAID dose (mg/day) | Effect on Cognitive Outcome Measures |
|---|---|---|---|---|---|---|---|---|---|
| Rogers et al. | 1993 | [65] | Mild to moderate AD | 44 | 28 | Indomethacin | 6 | 100–150 | Significant improvement |
| De Jong et al. | 2008 | [67] | Mild to moderate AD | 51 | 38 | Indomethacin | 12 | 100 | Not significant |
| Pasqualetti er al. | 2009 | [68] | Mild to moderate AD | 132 | 97 | Ibuprofen | 12 | 800 | Not significant |
| Aisen | 2002 | [71] | Mild to moderate AD | 40 | Nimesulide | 3 | 200 | Not significant | |
| Soininen | 2007 | [70] | Mild to moderate AD | 425 | 328 | Celecoxib | 12 | 400 | Not significant |
| Aisen et al. | 2003 | [66] | Mild to moderate AD | 351 | 351 | ||||
| Reines | 2004 | [69] | 692 | 481 | Rofecoxib | 12 | 25 | Not significant | |
| Martin | 2008 | [76] | Normal cognition >70 years; one relative with dementia | 2528 | 2117 | ||||
| Thal | 2005 | [72] | MCI | 1457 | 1457 | Rofecoxib | 48 | 25 | Significant impairment |
| Small | 2008 | [77] | age-associated memory decline | 88 | 40 | Cele | 18 | 200 or 400 | Significant improvement associated with increased glucose metabolism in prefrontal cortex |
5. Parkinson's Disease
5.1. Disease impact and pathology
5.2. Animal models investigating neuroinflammation in AD
5.3. Epidemiological observation of NSAIDs in PD incidence
5.4. Consideration of epidemiology to determine future investigation of NSAIDs in PD
6. NSAIDs in Other Neurological Disorders
7. Is There a Future for NSAIDs in Neurodegenerative Disease?
References
- Centers for Disease Control and Prevention, Health, United States; National Center for Health Statistics: Hyattsville, MD, USA, 2010.
- Alzheimer's Association. Alzheimer's disease Facts and Figures, 2010. Alzheimer's Dementia 2010, 6, 158–194. [CrossRef] [PubMed]
- Levine, D.B.; Fahrbach, K.R.; Siderowf, A.D.; Estok, R.P.; Ludensky, V.M.; Ross, S.D. Diagnosis and Treatment of Parkinson's Disease: A Systematic Review of the Literature; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2003. [Google Scholar]
- McGeer, P.L.; Schulzer, M.; McGeer, E.G. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: A review of 17 epidemiologic studies. Neurology 1996, 47, 425–432. [Google Scholar]
- Punchard, N.A.; Whelan, C.J.; Adcock, I. The Journal of Inflammation. J. Inflamm. 2004, 1, 1. [Google Scholar]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New. Biol. 1971, 231, 232–235. [Google Scholar]
- Kiefer, J.R.; Pawlitz, J.L.; Moreland, K.T.; Stegeman, R.A.; Hood, W.F.; Gierse, J.K.; Stevens, A.M.; Goodwin, D.C.; Rowlinson, S.W.; Marnett, L.J.; Stallings, W.C.; Kurumbail, R.G. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000, 405, 97–101. [Google Scholar]
- Hein, A.M.; O'Banion, M.K. Neuroinflammation and memory: The role of prostaglandins. Mol. Neurobio. 2009, 40, 15–32. [Google Scholar]
- Flower, R.J. The development of COX2 inhibitors. Nat. Rev. Drug Discov. 2003, 2, 179–191. [Google Scholar]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.T.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc. Nat. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar]
- Kis, B.; Snipes, J.A.; Gaspar, T.; Lenzser, G.; Tulbert, C.D.; Busija, D.W. Cloning of cyclooxygenase-1b (putative COX-3) in mouse. Inflamm. Res. 2006, 55, 274–278. [Google Scholar]
- McGeer, P.L. Cyclo-oxygenase-2 inhibitors: Rationale and therapeutic potential for Alzheimer's disease. Drugs Aging 2000, 17, 1–11. [Google Scholar]
- Breder, C.D.; Dewitt, D.; Kraig, R.P. Characterization of inducible cyclooxygenase in rat-brain. J. Comp. Neurol. 1995, 355, 296–315. [Google Scholar]
- Kaufmann, W.E.; Worley, P.F.; Pegg, J.; Bremer, M.; Isakson, P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc. Nat. Acad. Sci. USA 1996, 93, 2317–2321. [Google Scholar]
- Yamagata, K.; Andreasson, K.I.; Kaufmann, W.E.; Barnes, C.A.; Worley, P.F. Expression of a mitogen-inducible cyclooxygenase in brain neurons: Regulation by synaptic activity and glucocorticoids. Neuron 1993, 11, 371–386. [Google Scholar]
- Hein, A.M.; Stasko, M.R.; Matousek, S.B.; Scott-McKean, J.J.; Maier, S.F.; Olschowka, J.A.; Costa, A.C.S.; O'Banion, M.K. Sustained hippocampal IL-1β overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav. Immun. 2009, 24, 243–253. [Google Scholar]
- Moore, A.H.; Olschowka, J.A.; Williams, J.P.; Okunieff, P.; O'Banion, M.K. Regulation of prostaglandin E2 synthesis after brain irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 267–272. [Google Scholar]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 6th ed; Freeman and Company: New York, NY, USA, 2006. [Google Scholar]
- Marnett, L.J.; Rowlinson, S.W.; Goodwin, D.C.; Kalgutkar, A.S.; Lanzo, C.A. Arachidonic acid oxygenation by COX-1 and COX-2. Mechanisms of catalysis and inhibition. J. Biol. Chem. 1999, 274, 22903–22906. [Google Scholar] [PubMed]
- Cleland, L.G.; James, M.J. COX-2 selectivity varies across class. Med. J. Aust. 2005, 182, 197–198. [Google Scholar]
- Vane, J.R.; Botting, R.M. Anti-inflammatory drugs and their mechanism of action. Inflamm. Res. 1998, 47 Suppl. 2, S78–S87. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Streit, W.J. Microglia: Biology and pathology. Acta Neuropathol. 2010, 119, 89–105. [Google Scholar]
- Griffin, W.S.T.; Sheng, J.G.; Royston, M.C.; Gentleman, S.M.; McKenzie, J.E.; Graham, D.I.; Roberts, G.W.; Mrak, R.E. Glial-neuronal interactions in Alzheimer's disease: The potential role of a ''cytokine cycle'' in disease progression. Brain Pathol. 1998, 8, 65–72. [Google Scholar]
- Cacquevel, M.; Lebeurrier, N.; Cheenne, S.; Vivien, D. Cytokines in neuroinflammation and Alzheimer's disease. Curr. Drug Targets 2004, 5, 529–534. [Google Scholar]
- Shaftel, S.S.; Griffin, W.S.T.; O'Banion, M.K. The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J. Neuroinflamm. 2008, 5, 12. [Google Scholar]
- Cimino, P.J.; Keene, C.D.; Breyer, R.M.; Montine, K.S.; Montine, T.J. Therapeutic targets in prostaglandin E-2 signaling for neurologic disease. Curr. Med. Chem. 2008, 15, 1863–1869. [Google Scholar]
- Chen, C.; Bazan, N.G. Endogenous PGE(2) regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 2005, 93, 929–941. [Google Scholar]
- Chen, C.; Magee, J.C.; Bazan, N.G. Cyclooxygenase-2 regulates prostaglandin E-2 signaling in hippocampal long-term synaptic plasticity. J. Neurophysiol. 2002, 87, 2851–2857. [Google Scholar]
- Le, T.D.; Shirai, Y.; Okamoto, T.; Tatsukawa, T.; Nagao, S.; Shimizu, T.; Ito, M. Lipid signaling in cytosolic phospholipase A(2)alpha-cyclooxygenase-2 cascade mediates cerebellar long-term depression and motor learning. Proc. Nat. Acad. Sci. USA 2010, 107, 3198–3203. [Google Scholar]
- Savonenko, A.; Munoz, P.; Melnikova, T.; Wang, Q.; Liang, X.; Breyer, R.M.; Montine, T.J.; Kirkwood, A.; Andreasson, K. Impaired cognition, sensorimotor gating, and hippocampal long-term depression in mice lacking the prostaglandin E2 EP2 receptor. Exp. Neurol. 2009, 217, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Shie, F.S.; Montine, K.S.; Breyer, R.M.; Montine, T.J. Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia 2005, 52, 70–77. [Google Scholar]
- Takemiya, T.; Maehara, M.; Matsumura, K.; Yasuda, S.; Sugiura, H.; Yamagata, K. Prostaglandin E-2 produced by late induced COX-2 stimulates hippocampal neuron loss after seizure in the CA3 region. Neurosci. Res. 2006, 56, 103–110. [Google Scholar]
- Brown, G.C.; Bal-Price, A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol. Neurobiol. 2003, 27, 325–355. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Bates, T.E.; Stella, A.M.G. NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders: The role of oxidant/antioxidant balance. Neurochem. Res. 2000, 25, 1315–1341. [Google Scholar]
- Dawson, V.L.; Dawson, T.M. Nitric oxide in neurodegeneration. Prog. Brain Res. 1998, 118, 215–229. [Google Scholar]
- Parepally, J.M.R.; Mandula, H.; Smith, Q.R. Brain uptake of nonsteroidal anti-inflammatory drugs: Ibuprofen, flurbiprofen, and indomethacin. Pharmaceut. Res. 2006, 23, 873–881. [Google Scholar] [CrossRef]
- Plassman, B.L.; Langa, K.M.; Fisher, G.G.; Heeringa, S.G.; Weir, D.R.; Ofstedal, M.B.; Burke, J.R.; Hurd, M.D.; Potter, G.G.; Rodgers, W.L.; Steffens, D.C.; Willis, R.J.; Wallace, R.B. Prevalence of dementia in the united states: The aging, demographics, and memory study. Neuroepidemiology 2007, 29, 125–132. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Neurological Disorders: Public health challenges. Available online: http://www.who.int/mental_health/neurology/neurodiso/en/index.html (accessed June 01, 2010).
- Wimo, A.; Winblad, B.; Jonsson, L. An estimate of the total worldwide societal costs of dementia in 2005. Alzheimer. Dement. 2007, 3, 81–91. [Google Scholar]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; Finch, C.E.; Frautschy, S.; Griffin, W.S.; Hampel, H.; Hull, M.; Landreth, G.; Lue, L.; Mrak, R.; Mackenzie, I.R.; McGeer, P.L.; O'Banion, M.K.; Pachter, J.; Pasinetti, G.; Plata-Salaman, C.; Rogers, J.; Rydel, R.; Shen, Y.; Streit, W.; Strohmeyer, R.; Tooyoma, I.; Van Muiswinkel, F.L.; Veerhuis, R.; Walker, D.; Webster, S.; Wegrzyniak, B.; Wenk, G.; Wyss-Coray, T. Inflammation and Alzheimer's disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [PubMed]
- Small, G.W.; Agdeppa, E.D.; Kepe, V.; Satyamurthy, N.; Huang, S.C.; Barrio, J.R. In vivo brain imaging of tangle burden in humans. J. Mol. Neurosci. 2002, 19, 323–327. [Google Scholar] [PubMed]
- Small, G.W.; Bookheimer, S.Y.; Thompson, P.M.; Cole, G.M.; Huang, S.C.; Kepe, V.; Barrio, J.R. Current and future uses of neuroimaging for cognitively impaired patients. Lancet Neurol. 2008, 7, 161–172. [Google Scholar]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar]
- Rogers, J.; Lubernarod, J.; Styren, S.D.; Civin, W.H. Expression of immune system-associated antigens by cells of the human central nervous-system: Relationship to the pathology of alzheimers-disease. Neurobiol. Aging 1988, 9, 339–349. [Google Scholar]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer's disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar]
- Philipson, O.; Lord, A.; Gumucio, A.; O'Callaghan, P.; Lannfelt, L.; Nilsson, L.N.G. Animal models of amyloid-β-related pathologies in Alzheimer's disease. FEBS J. 2010, 277, 1389–1409. [Google Scholar]
- Meda, L.; Baron, P.; Scarlato, G. Glial activation in Alzheimer's disease: The role of Abeta and its associated proteins. Neurobiol. Aging 2001, 22, 885–893. [Google Scholar]
- Fujimi, K.; Noda, K.; Sasaki, K.; Wakisaka, Y.; Tanizaki, Y.; Iida, M.; Kiyohara, Y.; Kanba, S.; Iwaki, T. Altered expression of COX-2 in subdivisions of the hippocampus during aging and in Alzheimer's disease: The Hisayama study. Dement. Geriatr. Cogn. Disord. 2007, 23, 423–431. [Google Scholar]
- Hoozemans, J.J.M.; Rozemuller, J.M.; van Haastert, E.S.; Veerhuis, R.; Eikelenboom, P. Cyclooxygenase-1 and-2 in the different stages of Alzheimer's disease pathology. Curr. Pharm. Design 2008, 14, 1419–1427. [Google Scholar]
- Yermakova, A.V.; O'Banion, M.K. Downregulation of neuronal cyclooxygenase-2 expression in end stage Alzheimer's disease. Neurobiol. Aging 2001, 22, 823–836. [Google Scholar]
- Conde, J.R.; Streit, W.J. Microglia in the aging brain. J. Neuropathol. Exp. Neurol. 2006, 65, 199–203. [Google Scholar]
- Griffin, R.; Nally, R.; Nolan, Y.; McCartney, Y.; Linden, J.; Lynch, M.A. The age-related attenuation in long-term potentiation is associated with microglial activation. J. Neurochem. 2006, 99, 1263–1272. [Google Scholar]
- Murray, C.A.; Lynch, M.A. Evidence that increased hippocampal expression of the cytokine interleukin-1β is a common trigger for age- and stress-induced impairments in long-term potentiation. J. Neurosci. 1998, 18, 2974–2981. [Google Scholar]
- Perry, V.H.; Matyszak, M.K.; Fearn, S. Altered antigen expression of microglia in the aged rodent CNS. Glia 1993, 7, 60–67. [Google Scholar]
- Sheffield, L.G.; Berman, N.E.J. Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiol. Aging 1998, 19, 47–55. [Google Scholar]
- Hauss-Wegrzyniak, B.; Dobrzanski, P.; Stoehr, J.D.; Wenk, G.L. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer's disease. Brain Res. 1998, 780, 294–303. [Google Scholar]
- Hauss-Wegrzyniak, B.; Galons, J.P.; Wenk, G.L. Quantitative volumetric analyses of brain magnetic resonance imaging from rat with chronic neuroinflammation. Exp. Neurol. 2000, 165, 347–354. [Google Scholar]
- Moore, A.H.; Wu, M.; Shaftel, S.S.; Graham, K.A.; O'Banion, M.K. Sustained expression of interleukin-1β in mouse hippocampus impairs spatial memory. Neuroscience 2009, 164, 1484–1495. [Google Scholar]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar]
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; LaFerla, F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J. Neurosci. 2005, 25, 8843–8853. [Google Scholar]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar]
- Andreasson, K.I.; Savonenko, A.; Vidensky, S.; Goellner, J.J.; Zhang, Y.; Shaffer, A.; Kaufmann, W.E.; Worley, P.F.; Isakson, P.; Markowska, A.L. Age-dependent cognitive deficits and neuronal apoptosis in cyclooxygenase-2 transgenic mice. J. Neurosci. 2001, 21, 8198–8209. [Google Scholar]
- Hoshino, T.; Nakaya, T.; Homan, T.; Tanaka, K.I.; Sugimoto, Y.; Araki, W.; Narita, M.; Narumiya, S.; Suzuki, T.; Mizushima, T. Involvement of prostaglandin E-2 in production of amyloid-beta peptides both in vitro and in vivo. J. Biol. Chem. 2007, 282, 32676–32688. [Google Scholar] [PubMed]
- Liang, X.B.; Wang, Q.; Hand, T.; Wu, L.J.; Breyer, R.M.; Montine, T.J.; Andreasson, K. Deletion of the prostaglandin E-2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer's disease. J. Neurosci. 2005, 25, 10180–10187. [Google Scholar]
- Imbimbo, B.P. An update on the efficacy of non-steroidal anti-inflammatory drugs in Alzheimer's disease. Expert Opin. Investig. Drugs 2009, 18, 1147–1168. [Google Scholar]
- Lim, G.P.; Yang, F.; Chu, T.; Chen, P.; Beech, W.; Teter, B.; Tran, T.; Ubeda, O.; Ashe, K.H.; Frautschy, S.A.; Cole, G.M. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J. Neurosci. 2000, 20, 5709–5714. [Google Scholar]
- Yan, Q.; Zhang, J.H.; Liu, H.T.; Babu-Khan, S.; Vassar, R.; Biere, A.L.; Citron, M.; Landreth, G. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J. Neurosci. 2003, 23, 7504–7509. [Google Scholar]
- Stephan, A.; Laroche, S.; Davis, S. Learning deficits and dysfunctional synaptic plasticity induced by aggregated amyloid deposits in the dentate gyrus are rescued by chronic treatment with indomethacin. Eur. J. Neurosci. 2003, 17, 1921–1927. [Google Scholar]
- Cakala, M.; Malik, A.R.; Strosznajder, J.B. Inhibitor of cyclooxygenase-2 protects against amyloid beta peptide-evoked memory impairment in mice. Pharmacol. Rep. 2007, 59, 164–172. [Google Scholar]
- Rogers, J.; Kirby, L.C.; Hempelman, S.R.; Berry, D.L.; McGeer, P.L.; Kaszniak, A.W.; Zalinski, J.; Cofield, M.; Mansukhani, L.; Willson, P.; et al. Clinical trial of indomethacin in Alzheimer's disease. Neurology 1993, 43, 1609–1611. [Google Scholar]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J. Effects of rofecoxib or naproxen vs. placebo on Alzheimer disease progression: A randomized controlled trial. JAMA 2003, 289, 2819–2826. [Google Scholar] [PubMed]
- de Jong, D.; Jansen, R.; Hoefnagels, W.; Jellesma-Eggenkamp, M.; Verbeek, M.; Borm, G.; Kremer, B. No effect of one-year treatment with indomethacin on Alzheimer's disease progression: A randomized controlled trial. Plos One 2008, 3, e1475. [Google Scholar] [PubMed]
- Pasqualetti, P.; Bonomini, C.; Dal Forno, G.; Paulon, L.; Sinforiani, E.; Marra, C.; Zanetti, O.; Rossini, P.M. A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer's disease. Aging Clin. Exp. Res. 2009, 21, 102–110. [Google Scholar]
- Reines, S.A.; Block, G.A.; Morris, J.C.; Liu, G.; Nessly, M.L.; Lines, C.R.; Norman, B.A.; Baranak, C.C. Rofecoxib: No effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology 2004, 62, 66–71. [Google Scholar] [PubMed]
- Soininen, H.; West, C.; Robbins, J.; Niculescu, L. Long-term efficacy and safety of celecoxib in Alzheimer's disease. Dement. Geriatr. Cogn. Disord. 2007, 23, 8–21. [Google Scholar]
- Aisen, P.S.; Schmeidler, J.; Pasinetti, G.M. Randomized pilot study of nimesulide treatment in Alzheimer's disease. Neurology 2002, 58, 1050–1054. [Google Scholar]
- Thal, L.J.; Ferris, S.H.; Kirby, L.; Block, G.A.; Lines, C.R.; Yuen, E.; Assaid, C.; Nessly, M.L.; Norman, B.A.; Baranak, C.C.; Reines, S.A. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 2005, 30, 1204–1215. [Google Scholar] [PubMed]
- McGeer, P.L.; McGeer, E.G. NSAIDs and Alzheimer disease: Epidemiological, animal model and clinical studies. Neurobiol. Aging 2007, 28, 639–647. [Google Scholar]
- Townsend, K.P.; Pratico, D. Novel therapeutic opportunities for Alzheimer's disease: Focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005, 19, 1592–1601. [Google Scholar]
- Martin, B.K.; Szekely, C.; Brandt, J.; Piantadosi, S.; Breitner, J.C.S.; Craft, S.; Evans, D.; Green, R.; Mullan, M.; Grp, A.R. Cognitive function over time in the Alzheimer's disease anti-inflammatory prevention trial (ADAPT): Results of a randomized, controlled trial of naproxen and celecoxib. Arch. Neurol. 2008, 65, 896–905. [Google Scholar]
- Couzin, J. Clinical trials: Halt of Celebrex study threatens drug's future, other trials. Science 2004, 306, 2170–2170. [Google Scholar]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.N.; Johnson, R.E.; O'Banion, M.K. Sustained hippocampal IL-1β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J. Clin. Invest. 2007, 117, 1595–1604. [Google Scholar]
- Small, G.W.; Siddarth, P.; Silverman, D.H.; Ercoli, L.M.; Miller, K.J.; Lavretsky, H.; Bookheimer, S.Y.; Huang, S.C.; Barrio, J.R.; Phelps, M.E. Cognitive and cerebral metabolic effects of celecoxib vs. placebo in people with age-related memory loss: Randomized controlled study. Am. J. Geriatr. Psychiatry 2008, 16, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Rinne, J.O.; Tsui, W.H.; Berti, V.; Li, Y.; Wang, H.; Murray, J.; Scheinin, N.; Nagren, K.; Williams, S.; Glodzik, L.; De Santi, S.; Vallabhajosula, S.; de Leon, M.J. Increased fibrillar amyloid-β burden in normal individuals with a family history of late-onset Alzheimer's. Proc. Natl. Acad. Sci. USA 2010, 107, 5949–5954. [Google Scholar]
- Yang, H.W.; Zhang, J.; Breyer, R.M.; Chen, C. Altered hippocampal long-term synaptic plasticity in mice deficient in the PGE2 EP2 receptor. J. Neurochem. 2009, 108, 295–304. [Google Scholar]
- Cowley, T.R.; Fahey, B.; O'Mara, S.M. COX-2, but not COX-1, activity is necessary for the induction of perforant path long-term potentiation and spatial learning in vivo. Eur. J. Neurosci. 2008, 27, 2999–3008. [Google Scholar] [CrossRef] [PubMed]
- Guzman, C.B.; Graham, K.A.; Grace, L.A.; Moore, A.H. Sex-dependent effect of cyclooxygenase-2 inhibition on mouse spatial memory. Behav. Brain Res. 2009, 199, 355–359. [Google Scholar]
- Rall, J.M.; Mach, S.A.; Dash, P.K. Intrahippocampal infusion of a cyclooxygenase-2 inhibitor attenuates memory acquisition in rats. Brain Res. 2003, 968, 273–276. [Google Scholar]
- Sharifzadeh, M.; Naghdi, N.; Khosrovani, S.; Ostad, S.N.; Sharifzadeh, K.; Roghani, A. Post-training intrahippocampal infusion of the COX-2 inhibitor celecoxib impaired spatial memory retention in rats. Eur. J. Pharmacol. 2005, 511, 159–166. [Google Scholar]
- Teather, L.A.; Packard, M.G.; Bazan, N.G. Post-training cyclooxygenase-2 (COX-2) inhibition impairs memory consolidation. Learn. Memory 2002, 9, 41–47. [Google Scholar]
- Choi, S.H.; Aid, S.; Bosetti, F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: Implications for translational research. Trends Pharmacol. Sci. 2009, 30, 174–181. [Google Scholar]
- Szekely, C.A.; Green, R.C.; Breitner, J.C.S.; Ostbye, T.; Beiser, A.S.; Corrada, M.M.; Dodge, H.H.; Ganguli, M.; Kawas, C.H.; Kuller, L.H.; Psaty, B.M.; Resnick, S.M.; Wolf, P.A.; Zonderman, A.B.; Welsh-Bohmer, K.A.; Zandi, P.P. No advantage of Aβ42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology 2008, 70, 2291–2298. [Google Scholar]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar]
- Waldstein, S.R.; Wendell, C.R.; Seliger, S.L.; Ferrucci, L.; Metter, E.J.; Zonderman, A.B. Nonsteroidal anti-inflammatory drugs, Aspirin, and cognitive function in the Baltimore longitudinal study of aging. J. Amer. Geriat. Soc. 2010, 58, 38–43. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, H.S.; Shin, K.Y.; Kim, E.M.; Kim, M.; Park, C.H.; Jeong, Y.H.; Yoo, J.; Lee, J.P.; Chang, K.A.; Kim, S.; Suh, Y.H. Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer's disease models. Neuropsychopharmacology 2007, 32, 2393–2404. [Google Scholar]
- Noble, W.; Garwood, C.; Stephenson, J.; Kinsey, A.M.; Hanger, D.P.; Anderton, B.H. Minocycline reduces the development of abnormal tau species in models of Alzheimer's disease. FASEB J. 2009, 23, 739–750. [Google Scholar]
- Breitner, J.C.; Haneuse, S.J.; Walker, R.; Dublin, S.; Crane, P.K.; Gray, S.L.; Larson, E.B. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 2009, 72, 1899–1905. [Google Scholar]
- McKeage, K.; Blick, S.K.A.; Croxtall, J.D.; Lyseng-Williamson, K.A.; Keating, G.M. Esomeprazole: A review of its use in the management of gastric acid-related diseases in adults. Drugs 2008, 68, 1571–1607. [Google Scholar]
- Lleo, A.; Galea, E.; Sastre, M. Molecular targets of non-steroidal anti-inflammatory drugs in neurodegenerative diseases. Cell. Mol. Life Sci. 2007, 64, 1403–1418. [Google Scholar]
- Meinert, C.L.; McCaffrey, L.D.; Breitner, J.C. Alzheimer's disease anti-inflammatory prevention trial: Design, methods, and baseline results. Alzheimers Dement. 2009, 5, 93–104. [Google Scholar] [PubMed]
- Beher, D.; Clarke, E.E.; Wrigley, J.D.J.; Martin, A.C.L.; Nadin, A.; Churcher, I.; Shearman, M.S. Selected non-steroidal anti-inflammatory drugs and their derivatives target gamma-secretase at a novel site: Evidence for an allosteric mechanism. J. Biol. Chem. 2004, 279, 43419–43426. [Google Scholar]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O'Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar]
- Jantzen, P.T.; Connor, K.E.; DiCarlo, G.; Wenk, G.L.; Wallace, J.L.; Rojiani, A.M.; Coppola, D.; Morgan, D.; Gordon, M.N. Microglial activation and β-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J. Neurosci. 2002, 22, 2246–2254. [Google Scholar]
- Morihara, T.; Chu, T.; Ubeda, O.; Beech, W.; Cole, G.M. Selective inhibition of Aβ42 production by NSAID R-enantiomers. J. Neurochem. 2002, 83, 1009–1012. [Google Scholar]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Butler, T.; Kang, D.E.; Marquez-Sterling, N.; Golde, T.E.; Koo, E.H. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [Google Scholar]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. JAMA 2009, 302, 2557–2564. [Google Scholar]
- Masters, C. Anti-inflammatory drugs fall short in Alzheimer's disease. Nature Med. 2008, 14, 916. [Google Scholar]
- Hayden, K.M.; Zandi, P.P.; Khachaturian, A.S.; Szekely, C.A.; Fotuhi, M.; Norton, M.C.; Tschanz, J.T.; Pieper, C.F.; Corcoran, C.; Lyketsos, C.G.; Breitner, J.C.; Welsh-Bohmer, K.A. Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology 2007, 69, 275–282. [Google Scholar]
- Szekely, C.A.; Breitner, J.C.; Fitzpatrick, A.L.; Rea, T.D.; Psaty, B.M.; Kuller, L.H.; Zandi, P.P. NSAID use and dementia risk in the Cardiovascular Health Study: Role of APOE and NSAID type. Neurology 2008, 70, 17–24. [Google Scholar]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer's disease. Neuron 2009, 63, 287–303. [Google Scholar]
- Calabresi, P.; Picconi, B.; Parnetti, L.; Di Filippo, M. A convergent model for cognitive dysfunctions in Parkinson's disease: The critical dopamine-acetylcholine synaptic balance. Lancet Neurol. 2006, 5, 974–983. [Google Scholar]
- Lang, A.E.; Lozano, A.M. Parkinson's disease: First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar]
- Double, K.L.; Dedov, V.N.; Fedorow, H.; Kettle, E.; Halliday, G.M.; Garner, B.; Brunk, U.T. The comparative biology of neuromelanin and lipofuscin in the human brain. Cell. Mol. Life Sci. 2008, 65, 1669–1682. [Google Scholar]
- Fasano, M.; Giraudo, S.; Coha, S.; Bergamasco, B.; Lopiano, L. Residual substantia nigra neuromelanin in Parkinson's disease is cross-linked to α-synuclein. Neurochem. Int. 2003, 42, 603–606. [Google Scholar]
- Lang, A.E.; Lozano, A.M. Parkinson's disease: Second of two parts. N. Engl. J. Med. 1998, 339, 1130–1143. [Google Scholar]
- Searching for Answers: Families and Brain Disorder (Parkinson's Disease). Society for Neuroscience: Washington, DC, USA, 2009. Available online: http://www.sfn.org/index.cfm?pagename=SearchingforAnswers_FamiliesandBrainDisorders (accessed June 2nd, 2010).
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of parkinsons and alzheimers-disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson's disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar]
- Wilms, H.; Zecca, L.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Lucius, R. Inflammation in Parkinson's diseases and other neurodegenerative diseases: Cause and therapeutic implications. Curr. Pharm. Design 2007, 13, 1925–1928. [Google Scholar]
- Araujo, D.M.; Lapchak, P.A. Induction of immune-system mediators in the hippocampal-formation in alzheimers and parkinsons diseases: Selective effects on specific interleukins and interleukin receptors. Neuroscience 1994, 61, 745–754. [Google Scholar]
- Hunot, S.; Dugas, N.; Dugas, B.; Michel, P.P.; Faucheux, B.; Agid, Y.; Hirsch, E.C. Inflammatory mediators are expressed in the substantia nigra of patients with Parkinson's disease. Eur. J. Neurosci. 1998, 10, 6919. [Google Scholar]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1-β, interleukin-6, epidermal growth-factor and transforming growth-factor-α are elevated in the brain from parkinsonian-patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. tumor-necrosis-factor-α (TNF-α) increases both in the brain and in the cerebrospinal-fluid from parkinsonian-patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar]
- Wilms, H.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Zecca, L.; Lucius, R. Activation of microglia by human neuromelanin is NF-κB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson's disease. FASEB J. 2003, 17, 500–502. [Google Scholar]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with C-11 (R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar]
- Lima, M.D.S.; Reksidler, A.B.; Zanata, S.M.; Machado, H.B.; Tufik, S.; Vital, M. Different parkinsonism models produce a time-dependent induction of COX-2 in the substantia nigra of rats. Brain Res. 2006, 1101, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.M.; Kordower, J.H.; Collier, T.J. Age-related changes in glial cells of dopamine midbrain subregions in rhesus monkeys. Neurobiol. Aging 2008, 31, 937–952. [Google Scholar]
- Wang, T.G.; Pei, Z.; Zhang, W.; Liu, B.; Langenbach, R.; Lee, C.; Wilson, B.; Reece, J.M.; Miller, D.S.; Hong, J.S. MPP+-induced COX-2 activation and subsequent dopaminergic neurodegeneration. FASEB J. 2005, 19, 1134–1136. [Google Scholar]
- Teismann, P.; Tieu, K.; Choi, D.K.; Wu, D.C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc. Nat. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar]
- Feng, Z.H.; Wang, T.G.; Li, D.D.; Fung, P.; Wilson, B.C.; Liu, B.; Ali, S.F.; Langenbach, R.; Hong, J.S. Cyclooxygenase-2-deficient mice are resistant to 1-methyl-4-phenyl1,2,3,6-tetrahydropyridine-induced damage of dopaminergic neurons in the substantia nigra. Neurosci. Lett. 2002, 329, 354–358. [Google Scholar]
- Carrasco, E.; Casper, D.; Werner, P. Dopaminergic neurotoxicity by 6-OHDA and MPP+: Differential requirement for neuronal cyclooxygenase activity. J. Neurosci. Res. 2005, 81, 121–131. [Google Scholar]
- Carrasco, E.; Casper, D.; Werner, P. PGE(2) receptor EP1 renders dopaminergic neurons selectively vulnerable to low-level oxidative stress and direct PGE(2) neurotoxicity. J. Neurosci. Res. 2007, 85, 3109–3117. [Google Scholar]
- Carrasco, E.; Werner, P.; Casper, D. Prostaglandin receptor EP2 protects dopaminergic neurons against 6-OHDA-mediated low oxidative stress. Neurosci. Lett. 2008, 441, 44–49. [Google Scholar]
- Shie, F.S.; Montine, K.S.; Breyer, R.M.; Montine, T.J. Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia 2005, 52, 70–77. [Google Scholar]
- Jin, J.J.; Shie, F.S.; Liu, J.; Wang, Y.; Davis, J.; Schantz, A.M.; Montine, K.S.; Montine, T.J.; Zhang, J. Prostaglandin E-2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated α-synuclein. J. Neuroinflamm. 2007, 4, 10. [Google Scholar]
- Przedborski, S.; Goldman, J.E. Pathogenic role of glial cells in Parkinson's disease. Advan. Mol. Cell Biol. 2004, 31, 967–982. [Google Scholar]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson's disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Di Matteo, V.; Benigno, A.; Pierucci, M.; Crescimanno, G.; Di Giovanni, G. Non-steroidal anti-inflammatory drugs in Parkinson's disease. Exp. Neurol. 2007, 205, 295–312. [Google Scholar]
- Aubin, N.; Curet, O.; Deffois, A.; Carter, C. Aspirin and salicylate protect against MPTP-induced dopamine depletion in mice. J. Neurochem. 1998, 71, 1635–1642. [Google Scholar]
- Di Matteo, V.; Pierucci, M.; Di Giovanni, G.; Di Santo, A.; Poggi, A.; Benigno, A.; Esposito, E. Aspirin protects striatal dopaminergic neurons from neurotoxin-induced degeneration: An in vivo microdialysis study. Brain Res. 2006, 1095, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, H.; Maharaj, D.S.; Daya, S. Acetylsalicylic acid and acetaminophen protect against MPP+-induced mitochondrial damage and superoxide anion generation. Life Sci. 2006, 78, 2438–2443. [Google Scholar]
- Sairam, K.; Saravanan, K.S.; Banerjee, R.; Mohanakumar, K.P. Non-steroidal anti-inflammatory drug sodium salicylate, but not diclofenac or celecoxib, protects against 1-methyl-4-phenyl pyridinium-induced dopaminergic neurotoxicity in rats. Brain Res. 2003, 966, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Ferger, B. Inhibition of the cyclooxygenase isoenzymes COX-1 and COX-2 provide neuroprotection in the MPTP-mouse model of Parkinson's disease. Synapse 2001, 39, 167–174. [Google Scholar]
- Kurkowska-Jastrzebska, I.; Babiuch, M.; Joniec, I.; Przybylkowski, A.; Czlonkowski, A.; Czlonkowska, A. Indomethacin protects against neurodegeneration caused by MPTP intoxication in mice. Int. Immunopharmacol. 2002, 2, 1213–1218. [Google Scholar]
- Gupta, A.; Dhir, A.; Kumar, A.; Kulkarni, S.K. Protective effect of cyclooxygenase (COX)-inhibitors against drug-induced catatonia and MPTP-induced striatal lesions in rats. Pharmacol. Biochem. Behav. 2009, 94, 219–226. [Google Scholar]
- Klivenyi, P.; Gardian, G.; Calingasan, N.Y.; Yang, L.; Beal, M.F. Additive neuroprotective effects of creatine and a cyclooxygenase 2 inhibitor against dopamine depletion in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson's disease. J. Mol. Neurosci. 2003, 21, 191–198. [Google Scholar]
- Przybylkowski, A.; Kurkowska-Jastrzebska, I.; Joniec, I.; Ciesielska, A.; Czlonkowska, A.; Czlonkowski, A. Cyclooxygenases mRNA and protein expression in striata in the experimental mouse model of Parkinson's disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration to mouse. Brain Res. 2004, 1019, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Reksidler, A.B.; Lima, M.M.S.; Zanata, S.M.; Machado, H.B.; da Cunha, C.; Andreatini, R.; Tufik, S.; Vital, M. The COX-2 inhibitor parecoxib produces neuroprotective effects in MPTP-lesioned rats. Eur. J. Pharmacol. 2007, 560, 163–175. [Google Scholar]
- Sanchez-Pernaute, R.; Ferree, A.; Cooper, O.; Yu, M.; Brownell, A.L.; Isacson, O. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson's disease. J. Neuroinflamm. 2004, 1, 6. [Google Scholar]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Schwarzschild, M.A.; Willett, W.C.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 2003, 60, 1059–1064. [Google Scholar]
- Chen, H.L.; Jacobs, E.; Schwarzschild, M.A.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann. Neurol. 2005, 58, 963–967. [Google Scholar]
- Powers, K.M.; Kay, D.M.; Factor, S.A.; Zabetian, C.P.; Higgins, D.S.; Samii, A.; Nutt, J.G.; Griffith, A.; Leis, B.; Roberts, J.W.; Martinez, E.D.; Montimurro, J.S.; Checkoway, H.; Payami, H. Combined effects of smoking, coffee, and NSAIDs on Parkinson's disease risk. Movement Disord. 2008, 23, 88–95. [Google Scholar]
- Wahner, A.D.; Bronstein, J.M.; Bordelon, Y.M.; Ritz, B. Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology 2007, 69, 1836–1842. [Google Scholar]
- Bornebroek, M.; de Lau, L.M.L.; Haag, M.D.M.; Koudstaal, P.J.; Hofman, A.; Stricker, B.H.C.; Breteler, M.M.B. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Neuroepidemiology 2007, 28, 193–196. [Google Scholar]
- Bower, J.H.; Maraganore, D.M.; Peterson, B.J.; Ahlskog, J.E.; Rocca, W.A. Immunologic diseases, anti-inflammatory drugs, and Parkinson disease: A case-control study. Neurology 2006, 67, 494–496. [Google Scholar] [PubMed]
- Etminan, M.; Carleton, B.C.; Samii, A. Non-steroidal anti-inflammatory drug use and the risk of Parkinson disease: A retrospective cohort study. J. Clin. Neurosci. 2008, 15, 576–577. [Google Scholar]
- Etminan, M.; Suissa, S. NSAID use and the risk of Parkinson's disease. Curr. Drug Saf. 2006, 1, 223–225. [Google Scholar]
- Hernan, M.A.; Logroscino, G.; Garcia Rodriguez, L.A. Nonsteroidal anti-inflammatory drugs and the incidence of Parkinson disease. Neurology 2006, 66, 1097–1099. [Google Scholar]
- Gagne, J.J.; Power, M.C. Anti-inflammatory drugs and risk of Parkinson disease: A meta-analysis. Neurology 2010, 74, 995–1002. [Google Scholar]
- Samii, A.; Etminan, M.; Wiens, M.O.; Jafari, S. NSAID use and the risk of Parkinson's disease systematic review and meta-analysis of observational studies. Drug. Aging 2009, 26, 769–779. [Google Scholar]
- Gao, X.; Chen., H.; Schwarzschild, M.A.; Ascherio, A. Use of the non-steroidal anti-inflammatory drugs of Parkinson's disease: A prospective study and meta-analysis. In American Academy of Neurology 2010 Annual Meeting S03.003, Toronto, Ontario, Canada, April 17, 2010.
- Hirohata, M.; Ono, K.; Morinaga, A.; Yamada, M. Non-steroidal anti-inflammatory drugs have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. Neuropharmacology 2008, 54, 620–627. [Google Scholar] [CrossRef] [PubMed]
- O'Hare, E.; Elliott, J.J.; Hobson, P.; Spanswick, D.; Kim, E.M. Behavioural deterioration induced by intrahippocampal NAC61-95 injections and attenuation with ibuprofen. Behav. Brain Res. 2010, 208, 274–277. [Google Scholar]
- Kanaan, N.M.; Kordower, J.H.; Collier, T.J. Age and region-specific responses of microglia, but not astrocytes, suggest a role in selective vulnerability of dopamine neurons after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure in monkeys. Glia 2008, 56, 1199–1214. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, S.; Mori, A.; Kurihara, N.; Mitsumoto, Y.; Nakai, M. Age-related severity of dopaminergic neurodegeneration to MPTP neurotoxicity causes motor dysfunction in C57BL/6 mice. Neurosci. Lett. 2006, 401, 183–187. [Google Scholar]
- White, W.B.; West, C.R.; Borer, J.S.; Gorelick, P.B.; Lavange, L.; Pan, S.X.; Weiner, E.; Verburg, K.M. Risk of cardiovascular events in patients receiving celecoxib: A meta-analysis of randomized clinical trials. Amer. J. Cardiol. 2007, 99, 91–98. [Google Scholar]
- Becker, M.C.; Wang, T.H.; Wisniewski, L.; Wolski, K.; Libby, P.; Luscher, T.F.; Borer, J.S.; Mascette, A.M.; Husni, M.E.; Solomon, D.H.; Graham, D.Y.; Yeomans, N.D.; Krum, H.; Ruschitzka, F.; Lincoff, A.M.; Nissen, S.E.; Precision, I. Rationale, design, and governance of Prospective Randomized Evaluation of Celecoxib Integrated Safety vs. Ibuprofen Or Naproxen (PRECISION), a cardiovascular end point trial of nonsteroidal antiinflammatory agents in patients with arthritis. Amer. Heart J. 2009, 157, 606–612. [Google Scholar] [CrossRef]
- Hurley, S.D.; Olschowka, J.A.; O'Banion, M.K. Cyclooxygenase inhibition as a strategy to ameliorate brain injury. J. Neurotrauma 2002, 19, 1–15. [Google Scholar]
- Ahmad, M.; Zhang, Y.Q.; Liu, H.; Rose, M.E.; Graham, S.H. Prolonged opportunity for neuroprotection in experimental stroke with selective blockade of cyclooxygenase-2 activity. Brain Res. 2009, 1279, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E. Nimesulide as a promising neuroprotectant in brain ischemia: New experimental evidences. Pharmacol. Res. 2008, 57, 266–273. [Google Scholar]
- Dore, S.; Otsuka, T.; Mito, T.; Sugo, N.; Hand, T.; Wu, L.; Hurn, P.D.; Traystman, R.J.; Andreasson, K. Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Ann. Neurol. 2003, 54, 155–162. [Google Scholar]
- Khansari, P.S.; Halliwell, R.F. Evidence for neuroprotection by the fenamate NSAID, mefenamic acid. Neurochem. Int. 2009, 55, 683–688. [Google Scholar]
- Ladecola, C.; Niwa, K.; Nogawa, S.; Zhao, X.R.; Nagayama, M.; Araki, E.; Morham, S.; Ross, M.E. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc. Nat. Acad. Sci. USA 2001, 98, 1294–1299. [Google Scholar]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Lin, J.X.; Miyamoto, K.; Oka, N. A cyclooxygenase-2 inhibitor attenuates white matter damage in chronic cerebral ischemia. Neuroreport 1999, 10, 1461–1465. [Google Scholar]
- Bramlett, H.M.; Dietrich, W.D. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 2002, 103, 607–614. [Google Scholar]
- Dixon, C.E.; Kochanek, P.M.; Yan, H.Q.; Schiding, J.K.; Griffith, R.G.; Baum, E.; Marion, D.W.; DeKosky, S.T. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J. Neurotrauma 1999, 16, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, W.L.; Povlishock, J.T.; Graham, D.I. A mechanistic analysis of nondisruptive axonal injury: A review. J. Neurotrauma 1997, 14, 419–440. [Google Scholar]
- Nonaka, M.; Chen, X.H.; Pierce, J.E.S.; Leoni, M.J.; McIntosh, T.K.; Wolf, J.A.; Smith, D.H. Prolonged activation of NF-κB following traumatic brain injury in rats. J. Neurotrauma 1999, 16, 1023–1034. [Google Scholar]
- Evans, R.W.; Evans, R.I.; Sharp, M.J. The physician survey on the post-concussion and whiplash syndromes. Headache 1994, 34, 268–274. [Google Scholar]
- Ahmad, M.; Rose, M.E.; Vagni, V.; Griffith, R.P.; Dixon, C.E.; Kochanek, P.M.; Hickey, R.W.; Graham, S.H. Genetic disruption of cyclooxygenase-2 does not improve histological or behavioral outcome after traumatic brain injury in mice. J. Neurosci. Res. 2008, 86, 3605–3612. [Google Scholar]
- Kelso, M.L.; Scheff, S.W.; Pauly, J.R.; Loftin, C.D. Effects of genetic deficiency of cyclooxygenase-1 or cyclooxygenase-2 on functional and histological outcomes following traumatic brain injury in mice. BMC Neurosci. 2009, 10, 108. [Google Scholar]
- Cernak, I.; O'Connor, C.; Vink, R. Inhibition of cyclooxygenase 2 by nimesulide improves cognitive outcome more than motor outcome following diffuse traumatic brain injury in rats. Exp. Brain Res. 2002, 147, 193–199. [Google Scholar]
- Dash, P.K.; Mach, S.A.; Moore, A.N. Regional expression and role of cyclooxygenase-2 following experimental traumatic brain injury. J. Neurotrauma 2000, 17, 69–81. [Google Scholar]
- Kunz, T.; Marklund, N.; Hillered, L.; Oliw, E.H. Cyclooxygenase-2, prostaglandin synthases, and prostaglandin H-2 metabolism in traumatic brain injury in the rat. J. Neurotrauma 2002, 19, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Kelsen, J.; Kjaer, K.; Chen, G.; Pedersen, M.; Rohl, L.; Frokiaer, J.; Nielsen, S.; Nyengaard, J.R.; Ronn, L.C. Parecoxib is neuroprotective in spontaneously hypertensive rats after transient middle cerebral artery occlusion: A divided treatment response? J. Neuroinflamm. 2006, 3, 31. [Google Scholar] [CrossRef]
- Browne, K.D.; Iwata, A.; Putt, M.E.; Smith, D.H. Chronic ibuprofen administration worsens cognitive outcome following traumatic brain injury in rats. Exp. Neurol. 2006, 201, 301–307. [Google Scholar]
- Streit, W.J. Microglial senescence: Does the brain's immune system have an expiration date? Trends Neurosci. 2006, 29, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar]
- Ahmad, A.S.; Zhuang, H.; Echeverria, V.; Dore, S. Stimulation of prostaglandin EP2 receptors prevents NMDA-induced excitotoxicity. J. Neurotrauma 2006, 23, 1895–1903. [Google Scholar]
- Echeverria, V.; Clerman, A.; Dore, S. Stimulation of PGE receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following β-amyloid exposure. Eur. J. Neurosci. 2005, 22, 2199–2206. [Google Scholar]
- Groeger, A.L.; Cipollina, C.; Cole, M.P.; Woodcock, S.R.; Bonacci, G.; Rudolph, T.K.; Rudolph, V.; Freeman, B.A.; Schopfer, F.J. Cyclooxygenase-2 generates anti-inflammatory mediators from Ω-3 fatty acids. Nat. Chem. Biol. 2010, 6, 433–441. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moore, A.H.; Bigbee, M.J.; Boynton, G.E.; Wakeham, C.M.; Rosenheim, H.M.; Staral, C.J.; Morrissey, J.L.; Hund, A.K. Non-Steroidal Anti-Inflammatory Drugs in Alzheimer's Disease and Parkinson's Disease: Reconsidering the Role of Neuroinflammation. Pharmaceuticals 2010, 3, 1812-1841. https://doi.org/10.3390/ph3061812
Moore AH, Bigbee MJ, Boynton GE, Wakeham CM, Rosenheim HM, Staral CJ, Morrissey JL, Hund AK. Non-Steroidal Anti-Inflammatory Drugs in Alzheimer's Disease and Parkinson's Disease: Reconsidering the Role of Neuroinflammation. Pharmaceuticals. 2010; 3(6):1812-1841. https://doi.org/10.3390/ph3061812
Chicago/Turabian StyleMoore, Amy H., Matthew J. Bigbee, Grace E. Boynton, Colin M. Wakeham, Hilary M. Rosenheim, Christopher J. Staral, James L. Morrissey, and Amanda K. Hund. 2010. "Non-Steroidal Anti-Inflammatory Drugs in Alzheimer's Disease and Parkinson's Disease: Reconsidering the Role of Neuroinflammation" Pharmaceuticals 3, no. 6: 1812-1841. https://doi.org/10.3390/ph3061812
APA StyleMoore, A. H., Bigbee, M. J., Boynton, G. E., Wakeham, C. M., Rosenheim, H. M., Staral, C. J., Morrissey, J. L., & Hund, A. K. (2010). Non-Steroidal Anti-Inflammatory Drugs in Alzheimer's Disease and Parkinson's Disease: Reconsidering the Role of Neuroinflammation. Pharmaceuticals, 3(6), 1812-1841. https://doi.org/10.3390/ph3061812