Phospholipase D2 Enhances Epidermal Growth Factor-Induced Akt Activation in EL4 Lymphoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
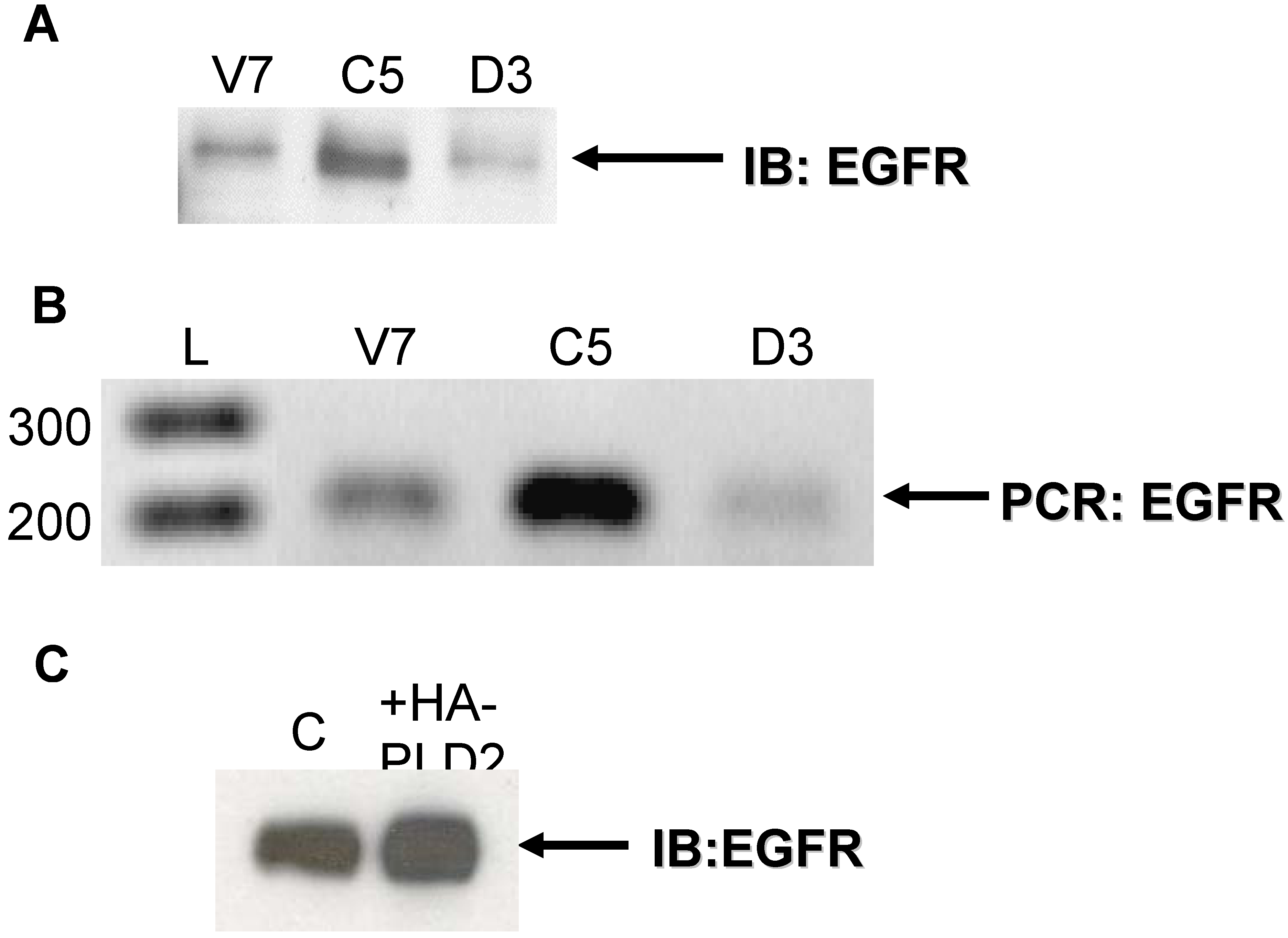
2.1. Expression of EGFR in EL4 Lymphoma Cells

2.2. Effects of Over-Expression of PLD2 in OVCAR3 Ovarian Cancer Cells
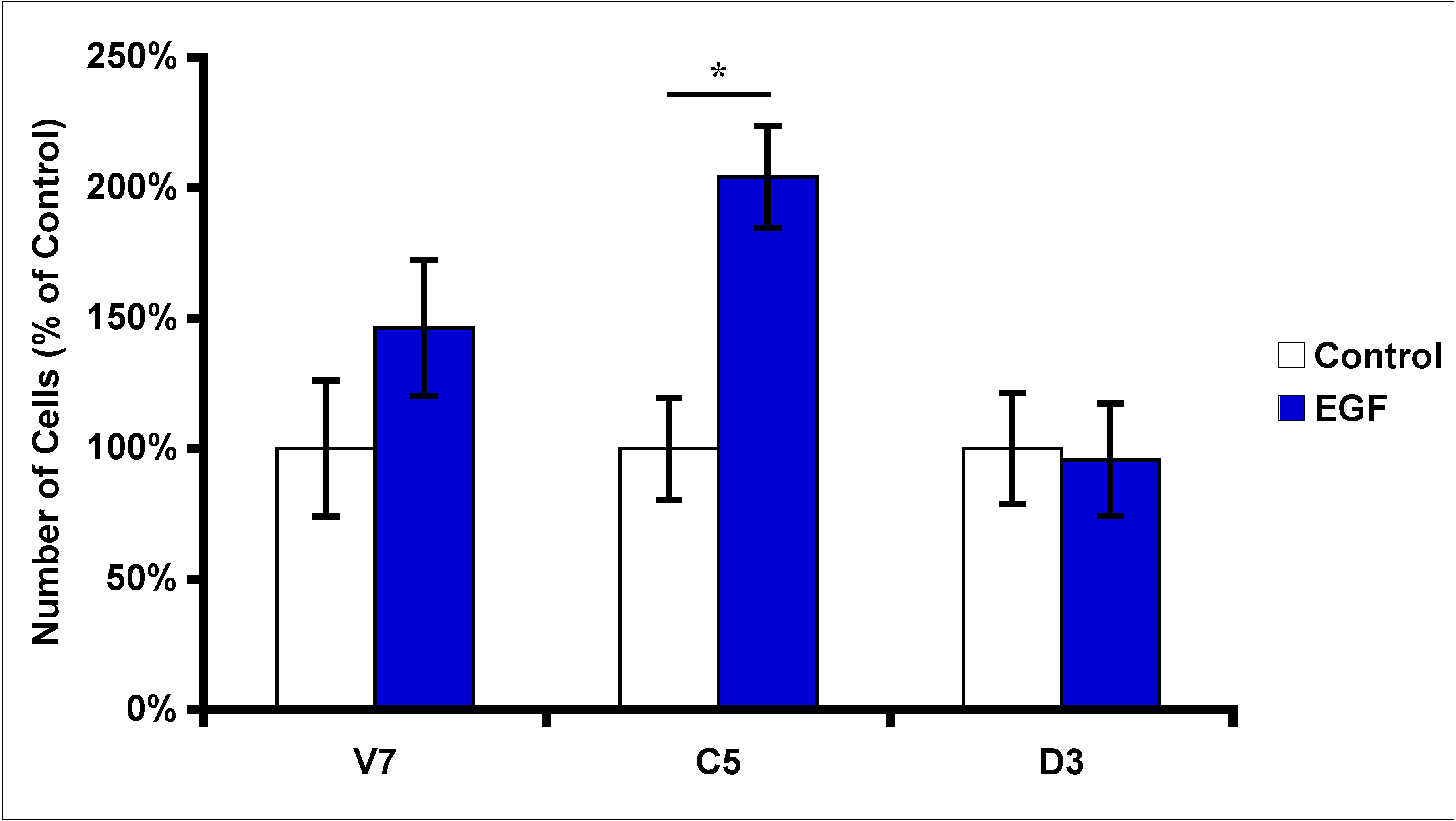
2.3. Effects of EGF Treatment on EL4 Cell Proliferation

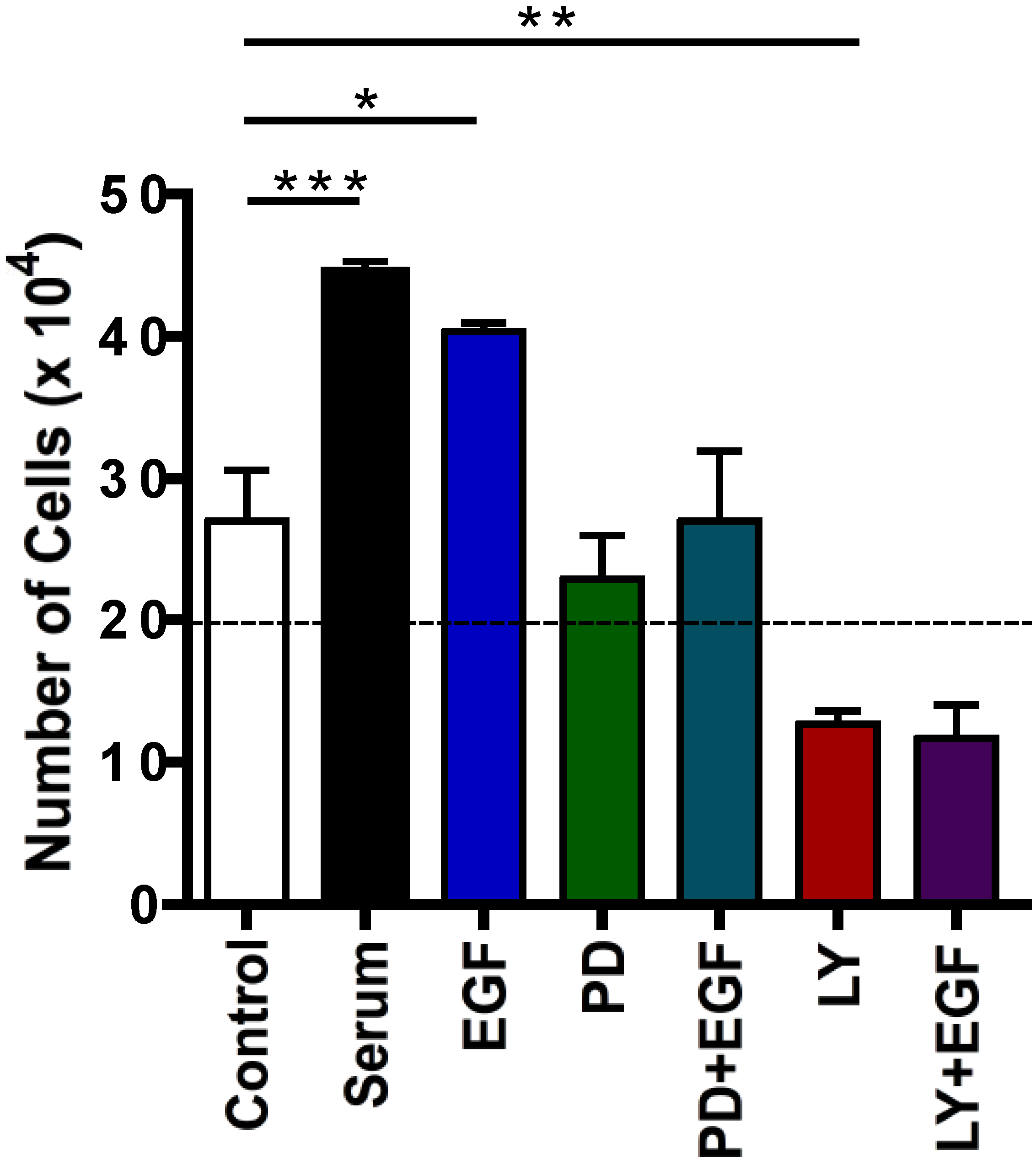
2.4. Effects of Protein Kinase Inhibitors on EGF-Induced C5 Cell Proliferation

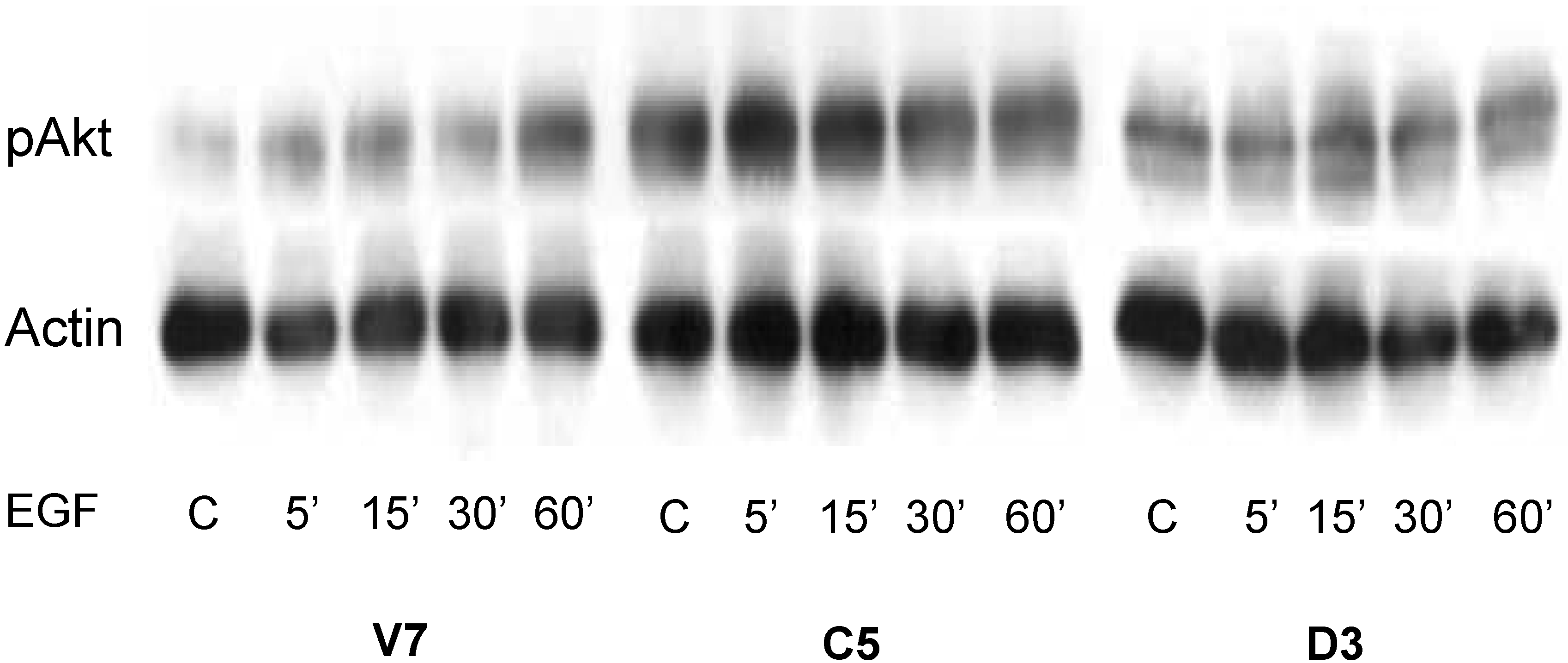
2.5. Time Course of EGF-Induced Akt Activation in EL4 Cells

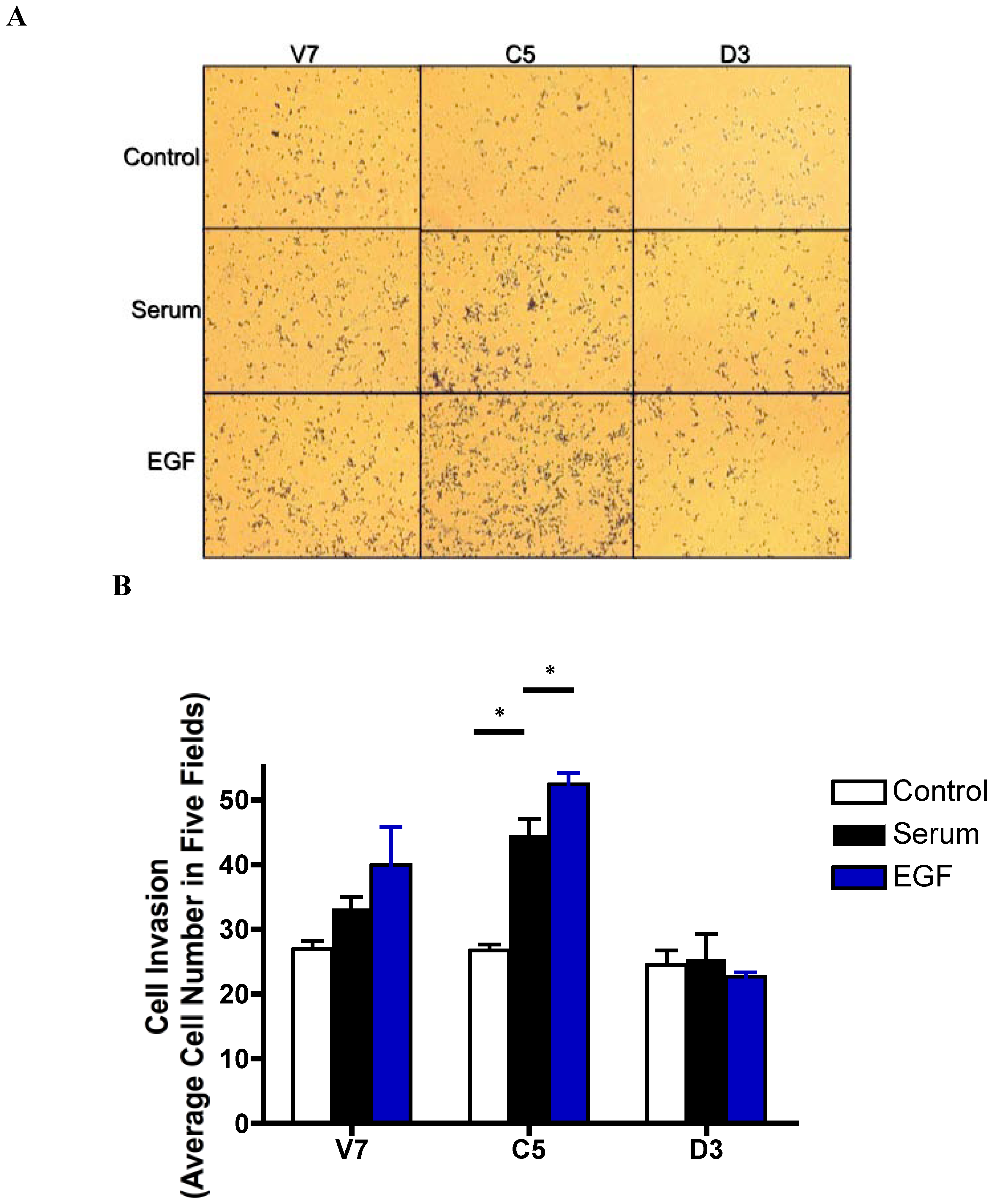
2.6. Effects of EGF-Induced Cell Invasion in EL4 Cells

2.7. Discussion
3. Experimental Section
3.1. Materials
3.2. Cell Culture
3.3. Immunoblotting
3.4. Semi-Quantitative PCR
3.5. PLD2 Transfection
3.6. Cell Proliferation
3.7. Cell Invasion Assay
3.8. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Banno, Y. Regulation and possible role of mammalian phospholipase D in cellular functions. J. Biochem. 2002, 131, 301–306. [Google Scholar]
- Foster, D.A.; Xu, L. Phospholipase D in cell proliferation and cancer. Mol. Cancer Res. 2003, 1, 789–800. [Google Scholar]
- Meier, K.E.; Gibbs, T.C.; Knoepp, S.M.; Ella, K.M. Expression of phospholipase D isoforms in mammalian cells. Biochem. Biophys. Acta. 1999, 1439, 199–213. [Google Scholar]
- McDermott, M.; Wakelam, M.J.; Morris, A.J. Phospholipase D. Biochem. Cell Biol. 2004, 82, 225–253. [Google Scholar]
- Powner, D.J.; Wakelam, M.J. The regulation of phospholipase D by inositol phospholipids and small GTPases. FEBS Lett. 2002, 531, 62–64. [Google Scholar]
- Exton, J.H. Regulation of phospholipase D. FEBS Lett. 2002, 531, 58–61. [Google Scholar]
- Liscovitch, M.; Czarny, M.; Flucci, G.; Lavie, Y.; Tang, X. Localization and possible functions of phospholipase D isozymes. Biochim. Biophys. Acta. 1999, 1439, 245–263. [Google Scholar]
- Ktistakis, N.T.; Delon, C.; Manifava, M.; Wood, E.; Ganley, I.; Sugars, J.M. Phospholipase D1 and potential targets of its hydrolysis product, phosphatidic acid. Biochem. Soc. Trans. 2003, 31, 94–97. [Google Scholar] [PubMed]
- O'Luanaigh, N.; Pardo, R.; Fensome, A.; Allen-Baume, V.; Jones, D.; Holt, M.R.; Cockcroft, S. Continual production of phosphatidic acid by phospholipase D is essential for antigen-stimulated membrane ruffling in cultured mast cells. Mol. Biol. Cell. 2002, 13, 3730–3746. [Google Scholar]
- Lehman, N.; Ledford, B.; Fulvio, M.; Frondorf, K.; McPhail, L.C.; Gomez-Cambronero, J. Phospholipase D2-derived phosphatidic acid binds to and activates ribosomal p70 S6 kinase independently of mTOR. FASEB J. 2007, 21, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Jang, I.H.; Lee, S.; Park, J.B.; Kim, J.H.; Lee, C.S.; Hur, E.M.; Kim, I.S.; Kim, K.T.; Yagisawa, H.; Suh, P.G.; Ryu, S.H. The direct interaction of phospholipase C-g1 with phospholipase D2 is important for epidermal growth factor signaling. J. Biol. Chem. 2003, 278, 18184–18190. [Google Scholar]
- Yang, L.Q.; Seifert, A.; Wu, D.F.; Wang, X.; Rankovic, V.; Schroeder, H.; Brandenberg, L.O.; Hoellt, V.; Koch, T. Role of phospholipase D2/phosphatidic acid signal transduction in µ- and d-opioid receptor endocytosis. Mol. Pharmacol. 2010. [Google Scholar]
- Luquain, C.; Singh, A.; Wang, L.; Natarajan, V.; Morris, A.J. Role of phospholipase D in agonist-stimulated lysophosphatidic acid synthesis by ovarian cancer cells. J. Lipid Res. 2003, 44, 1963–1975. [Google Scholar]
- Lee, J.H.; Kim, Y.M.; Ki, N.W.; Kim, J.W.; Her, E.; Kim, B.K.; Kim, J.H.; Ryu, S.H.; Park, J.W.; Seo, D.W.; Han, J.W.; Beaven, M.A.; Choi, W.S. Phospholipase D2 acts as an essential adaptor protein in the activation of Syk in antigen-stimulated mast cells. Blood 2006, 108, 956–964. [Google Scholar]
- Knoepp, S.M.; Chahal, M.S.; Xie, Y.; Zhang, Z.; Brauner, D.J.; Hallman, M.A.; Robinson, S.A.; Han, S.; Imai, M.; Tomlinson, S.; Meier, K.E. Effects of active and inactive phospholipase D2 on signal transduction, adhesion, migration, invasion, and metastasis in EL4 lymphoma cells. Mol. Pharmacol. 2008, 74, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Rodrik, V.; Foster, D.A. Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene 2005, 24, 672–679. [Google Scholar]
- Zhao, Y.; Ehara, H.; Akao, Y.; Shamoto, M.; Nakagawa, Y.; Banno, Y.; Deguchi, T.; Ohishi, N.; Yagi, K.; Nozawa, Y. Increased activity and intranuclear expression of phospholipase D2 in human renal cancer. Biochem. Biophys. Res. Commun. 2000, 278, 140–143. [Google Scholar]
- Rodriquez-Gonzalez, A.; Ramirez de Molina, A.; Benitez-Rajal, J.; Lacal, J.C. Phospholipase D and choline kinase: their role in cancer development and their potential as drug targets. Prog. Cell Cycle Res. 2003, 5, 191–201. [Google Scholar]
- Joseph, T.; Bryant, A.; Frankel, P.; Wooden, R.; Kerkhoff, E.; Rapp, U.R.; Foster, D.A. Phospholipase D overcomes cell cycle arrest induced by high-intensity Raf signaling. Oncogene 2002, 16, 3651–3658. [Google Scholar]
- Chen, J.; Fang, Y. A novel pathway regulating the mammalian target of rapamycin (mTOR) signaling. Biochem. Pharmacol. 2002, 64, 1071–1077. [Google Scholar]
- Zhong, M.; Joseph, T.; Jackson, D.; Beychenok, S.; Foster, D.A. Elevated phospholipase D activity induces apoptosis in normal rat fibroblasts. Biochem. Biophys. Res. Commun. 2002, 298, 474–477. [Google Scholar]
- Yamada, Y.; Hamajima, N.; Kato, T.; Iwata, H.; Yamamura, Y.; Shinoda, M.; Suyama, M.; Mitsudomi, T.; Tajima, K.; Kusakabe, S.; Yoshida, H.; Banno, Y.; Akao, Y.; Tanaska, M.; Nozawa, Y. Association of a polymorphism of the phospholipase D2 gene with the prevalence of colorectal cancer. J. Mol. Med. 2003, 81, 126–131. [Google Scholar]
- Xu, L.; Shen, Y.; Joseph, T.; Bryant, A.; Luo, J.Q.; Frankel, P.; Rotunda, T.; Foster, D.A. Mitogenic phospholipase D activity is restricted to caveolin-enriched membrane microdomains. Biochem. Biophys. Res. Commun. 2000, 273, 77–83. [Google Scholar]
- Cabodi, S.; Moro, L.; Bergatto, E.; Boeri Erba, E.; Di Stefano, P.; Turco, E.; Tarone, G.; Defilippi, P. Integrin regulation of epidermal growth factor (EGF) receptor and of EGF-dependent responses. Biochem. Soc. Trans. 2004, 32, 438–442. [Google Scholar]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgress, A.W. Epidermal growth factor receptor: Mechanisms of activation and signaling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar]
- Barnes, C.J.; Kumar, R. Biology of the epidermal growth factor receptor family. Cancer Treat. Res. 2004, 119, 1–13. [Google Scholar]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar]
- Sundaram, M.; Cook, H.W.; Byers, D.M. The MARCKS family of phospholipid binding proteins: Regulation of phospholipase D and other cellular components. Biochem. Cell Biol. 2004, 82, 191–200. [Google Scholar]
- Kim, S.W.; Hayashi, M.; Lo, J.F.; Yang, Y.; Yoo, J.S.; Lee, J.D. ADP-ribosylation factor 4 small GTPase mediates epidermal growth factor receptor-dependent phospholipase D2 activation. J. Biol. Chem. 2003, 278, 2661–2668. [Google Scholar]
- Lee, H.Y.; Park, J.B.; Jang, I.H.; Chae, Y.C.; Kim, J.H.; Kim, I.S.; Suh, P.G.; Ryu, S.H. Munc-18-1 inhibits phospholipase D activity by direct interaction in an epidermal growth factor-reversible manner. J. Biol. Chem. 2004, 279, 16339–16348. [Google Scholar]
- Reiter, J.L.; Threadgill, D.W.; Eley, G.D.; Strunk, K.E.; Danielsen, A.J.; Sinclair, C.S.; Pearsall, R.S.; Green, P.J.; Yee, D.; Lampland, A.L.; Balasubramaniam, S.; Crossley, T.D.; Magnuson, T.R.; James, C.D.; Malhle, N.J. Comparative genomic sequence analysis and isolation of human and mouse alternative EGFR transcripts encoding truncated receptor isoforms. Genomics 2001, 71, 1–20. [Google Scholar]
- Snider, A.J.; Meier, K.E. Receptor transactivation cascades. Am. J. Physiol. Cell. Physiol. 2007, 292, C1–C3. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.; Meier, K.E. Phosphorylation of paxillin via the ERK mitogen-activated protein kinase cascade in EL4 thymoma cells. J. Biol. Chem. 275, 15, 11333–11340. [Google Scholar]
- Han, S.; Knoepp, S.M.; Hallman, M.A.; Meier, K.E. RasGRP1 confers the phorbol ester-sensitive phenotype to EL4 lymphoma cells. Mol. Pharmacol. 2007, 71, 314–322. [Google Scholar]
- Van Kolen, K.; Gilany, K.; Moens, L.; Esmans, E.L.; Slegers, H. P2Y12 receptor signaling towards PKB proceeds through IGF-1 receptor cross-talk and activation of Src, Pyk2 and Rap1. Cell Signal. 2008, 18, 1169–1181. [Google Scholar]
- Bor, M.V.; Sorensen, B.S.; Rammer, P.; Nexo, E. Calibrated user-friendly reverse transcriptase-PCR assay: Quantitation of epidermal growth factor receptor mRNA. Clin. Chem. 1998, 44, 1154–1160. [Google Scholar]
- Rehman, A.; Taishi, P.; Fang, J.; Majde, J.A.; Krueger, J.M. The cloning of a rat peptidoglycan recognition protein (PGRP) and its induction in brain by sleep deprivation. Cytokine 2001, 13, 8–17. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chahal, M.S.; Brauner, D.J.; Meier, K.E. Phospholipase D2 Enhances Epidermal Growth Factor-Induced Akt Activation in EL4 Lymphoma Cells. Pharmaceuticals 2010, 3, 2045-2058. https://doi.org/10.3390/ph3072045
Chahal MS, Brauner DJ, Meier KE. Phospholipase D2 Enhances Epidermal Growth Factor-Induced Akt Activation in EL4 Lymphoma Cells. Pharmaceuticals. 2010; 3(7):2045-2058. https://doi.org/10.3390/ph3072045
Chicago/Turabian StyleChahal, Manpreet S., Daniel J. Brauner, and Kathryn E. Meier. 2010. "Phospholipase D2 Enhances Epidermal Growth Factor-Induced Akt Activation in EL4 Lymphoma Cells" Pharmaceuticals 3, no. 7: 2045-2058. https://doi.org/10.3390/ph3072045
APA StyleChahal, M. S., Brauner, D. J., & Meier, K. E. (2010). Phospholipase D2 Enhances Epidermal Growth Factor-Induced Akt Activation in EL4 Lymphoma Cells. Pharmaceuticals, 3(7), 2045-2058. https://doi.org/10.3390/ph3072045