Statistical Estimation of the Protein-Ligand Binding Free Energy Based On Direct Protein-Ligand Interaction Obtained by Molecular Dynamics Simulation

Abstract
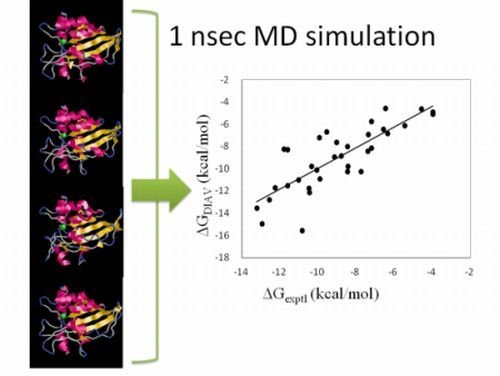
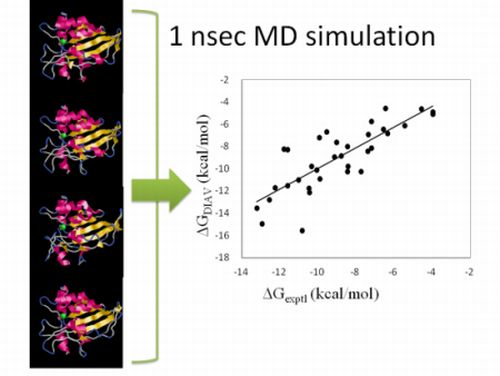
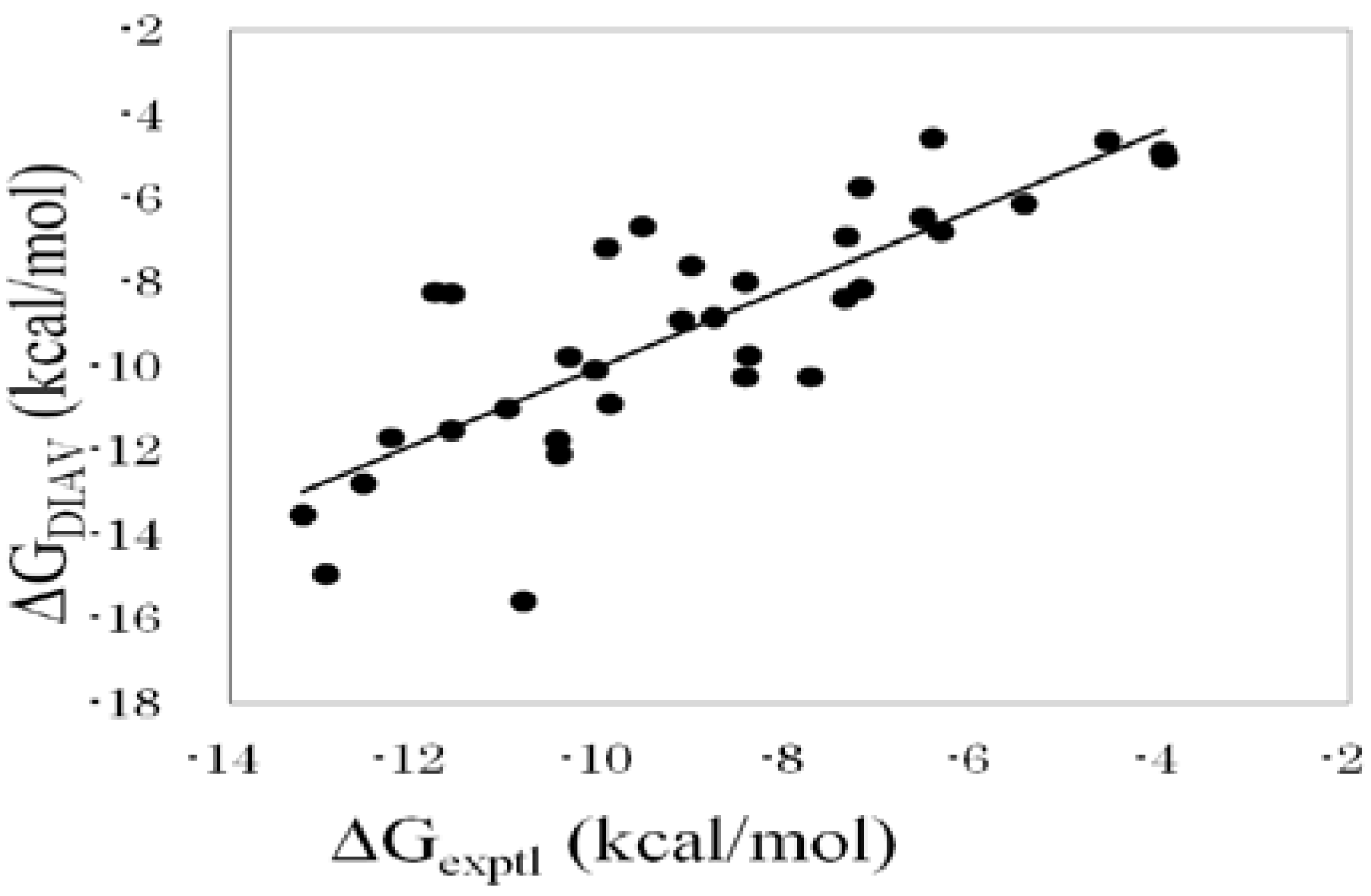
:
1. Introduction

2. Results and Discussion
2.1. Theoretical Background









2.2. Entropy Term

2.3. Modification of van der Waals Potential Term




2.4. Effective Dielectric Constant





2.5. Examination of Entropy Term

{kind=link}
{kind=link}
| Statistics | ΔGsimple (Equation 10) | ELE a | vdW a | ASA a | DIH a |
|---|---|---|---|---|---|
| Average error (kcal/mol) | 2.22 | 2.30 | 1.85 | 2.06 | 1.94 |
| R | 0.72 | 0.70 | 0.72 | 0.71 | 0.67 |
| α | 0.22 | 0.22 | 0.18 | 0.17 | 0.15 |
| β | 0.017001 | 0.016411 | 0.005958 | 0.012600 | 0.010430 |
| τ*100 | - | −0.078460 | −28.506610 | −0.026605 | −0.000696 |
2.6. Examination of vdW Term
| Statistics | LJ9-6 | LJ8-4 | LJ6-3 |
|---|---|---|---|
| Average error (kcal/mol) | 2.26 | 1.75 | 1.89 |
| R | 0.69 | 0.76 | 0.71 |
| α | 0.1727 | 0.0428 | 0.0066 |
| β | 0.0139 | 0.0072 | 0.0078 |
| τ*10000 | −2.9273 | −2.5677 | −2.8531 |
2.7. Examination of α2 and β2 Parameters
2.8. Examination of Effective Dielectric Constant Term
| Statistics | ASA | DIH |
|---|---|---|
| Average error (kcal/mol) | 1.63 | 1.59 |
| R | 0.80 | 0.76 |
| α | 0.04146 | 0.03832 |
| β | 0.00643 | 0.00491 |
| τ*10000 | −2.74887 | −0.06949 |
| α2 | 0.0093 | 0.0093 |
| β2 | −0.0013 | −0.0015 |
| PDB ID | ΔGexptl (kcal/mol) | ΔGsimple (Equation (10)) (kcal/mol) | ΔGDIAV (Equation (11)) (kcal/mol) | ΔGDIAS (Equation (16)) (kcal/mol) |
|---|---|---|---|---|
| 1abe | −9.57 | −5.46 | −6.27 | −6.68 |
| 1abf | −7.39 | −6.30 | −6.67 | −6.90 |
| 1apu | −10.50 | −13.50 | −11.98 | −11.76 |
| 1dbb | −12.27 | −8.75 | −11.79 | −11.69 |
| 1dbj | −10.47 | −8.35 | −12.27 | −12.10 |
| 1dog | −5.48 | −5.40 | −6.09 | −6.12 |
| 1dwb | −3.98 | −3.69 | −4.83 | −5.05 |
| 1epo | −10.85 | −17.25 | −14.82 | −15.56 |
| 1etr | −10.09 | −9.91 | −10.35 | −10.08 |
| 1ets | −11.62 | −11.05 | −11.82 | −11.52 |
| 1ett | −8.44 | −9.46 | −9.99 | −9.75 |
| 1hpv | −12.57 | −14.02 | −12.88 | −12.78 |
| 1hsl | −9.96 | −6.53 | −6.74 | −7.18 |
| 1htf | −11.04 | −12.45 | −11.12 | −11.00 |
| 1hvr | −12.97 | −16.98 | −14.67 | −14.95 |
| 1nsd | −7.23 | −7.44 | −8.33 | −8.13 |
| 1pgp | −7.77 | −11.01 | −11.09 | −10.24 |
| 1phg | −11.81 | −6.88 | −8.03 | −8.22 |
| 1ppc | −8.80 | −9.83 | −8.66 | −8.85 |
| 1pph | −8.49 | −8.50 | −7.87 | −8.00 |
| 1rbp | −9.17 | −9.29 | −8.58 | −8.91 |
| 1tng | −4.00 | −4.15 | −4.64 | −4.90 |
| 1tnh | −4.59 | −3.54 | −4.24 | −4.61 |
| 1ulb | −7.23 | −3.82 | −5.71 | −5.74 |
| 2cgr | −9.92 | −7.07 | −10.94 | −10.88 |
| 2gbp | −10.36 | −8.95 | −9.27 | −9.77 |
| 2ifb | −7.41 | −9.57 | −8.53 | −8.38 |
| 2phh | −6.38 | −4.09 | −6.83 | −6.79 |
| 2r04 | −8.48 | −10.39 | −10.31 | −10.26 |
| 2tsc | −11.62 | −11.05 | −8.68 | −8.28 |
| 2ypi | −6.58 | −5.40 | −5.72 | −6.45 |
| 3ptb | −6.46 | −4.93 | −5.02 | −4.55 |
| 4dfr | −13.23 | −11.52 | −13.93 | −13.52 |
| 5abp | −9.05 | −6.64 | −7.19 | −7.59 |
| Averageerror | - | 1.88 | 1.30 | 1.22 |
| R | - | 0.73 | 0.81 | 0.81 |
| α | - | 0.0503 | 0.0378 | 0.0307 |
| β | - | 0.0125 | 0.0082 | 0.0118 |
| τ∗10000 | - | - | −2.4178 | −2.4312 |
| α2 | - | - | 0.0093 | 0.01 |
| β2 | - | - | −0.0011 | −0.00312 |
| x | - | - | - | 0.6 |


2.9. Application to Docking-Pose Prediction

| Initial structure (intact PDB coordinates: model 1) | Top ΔG structure by the DIAS method | Top scoring structure by Sievgene | Best among the top 5 structures |
|---|---|---|---|
| RMSD < 1 Å | 29.4% | 35.3% | 47.1% |
| RMSD < 2 Å | 41.2% | 76.5% | 94.1% |
| RMSD < 3 Å | 47.1% | 94.1% | 94.1% |
| Energy-minimized structure (model 2) | Top ΔG structure by the DIAS method | Top scoring structure by Sievgene | Best among the top 5 structures |
| RMSD < 1 Å | 40.0% | 6.7% | 66.7% |
| RMSD < 2 Å | 73.3% | 46.7% | 93.3% |
| RMSD < 3 Å | 80.0% | 73.3% | 93.3% |
| Structure after MD simulation (model 3) | Top ΔG structure by the DIAS method | Top scoring structure by Sievgene | Best among the top 5 structures |
| RMSD < 1 Å | 20.0% | 0.0% | 0.0% |
| RMSD < 2 Å | 33.3% | 33.3% | 33.3% |
| RMSD < 3 Å | 53.3% | 46.7% | 66.7% |
3. Data Preparation
| PDB ID | Protein |
|---|---|
| 1abe | L-ARABINOSE-BINDING PROTEIN |
| 1abf | L-ARABINOSE-BINDING PROTEIN |
| 1apu | ACID PROTEINASE (PENICILLOPEPSIN) |
| 1dbb | FAB' FRAGMENT |
| 1dbj | FAB' FRAGMENT |
| 1dog | GLUCOAMYLASE |
| 1dwb | THROMBIN |
| 1epo | ENDOTHIA ASPARTIC PROTEINASE |
| 1etr | THROMBIN |
| 1ets | THROMBIN |
| 1ett | THROMBIN |
| 1hpv | HIV-1 PROTEASE |
| 1hsl | HISTIDINE-BINDING PROTEIN |
| 1htf | HIV-1 PROTEASE |
| 1hvr | HIV-1 PROTEASE |
| 1nsd | NEURAMINIDASE |
| 1pgp | 6-PHOSPHOGLUCONATE DEHYDROGENASE |
| 1phg | CYTOCHROME P450 |
| 1ppc | TRYPSIN |
| 1pph | TRYPSIN |
| 1rbp | RETINOL-BINDING PROTEIN |
| 1tng | TRYPSIN |
| 1tnh | TRYPSIN |
| 1ulb | PURINE NUCLEOSIDE PHOSPHORYLASE |
| 2cgr | IGG2B (KAPPA) FAB FRAGMENT |
| 2gbp | D-GALACTOSE/D-GLUCOSE-BINDING PROTEIN |
| 2ifb | INTESTINAL FATTY ACID BINDING |
| 2phh | P-HYDROXYBENZOATE HYDROXYLASE |
| 2r04 | RHINOVIRUS 14 (HRV14) |
| 2tsc | THYMIDYLATE SYNTHASE |
| 2ypi | TRIOSE PHOSPHATE ISOMERASE |
| 3ptb | TRYPSIN |
| 4dfr | DIHYDROFOLATE REDUCTASE |
| 5abp | L-ARABINOSE-BINDING PROTEIN |
4. Conclusions
Acknowledgements
References
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]
- Kontoyianni, M.; Sokol, G.S.; McClellan, L.M. Evaluation of library ranking efficacy in virtual screening. J. Comput. Chem. 2005, 26, 11–22. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef]
- Jones, G.; Willet, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Baxter, C.A.; Murray, C.W.; Clark, D.E.; Westhead, D.R.; Eldridge, M.D. Flexible docking using tabu search and an empirical estimate of binding affinity. Proteins 1998, 33, 367–382. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Mikami, Y.; Nakamura, H. Similarities among receptor pockets and among compounds: Analysis and application to in silico ligand screening. J. Mol. Graph. Model. 2005, 24, 34–45. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, S.; Zhu, Q.; Zhou, Y. A knowledge-based energy function for protein-ligand, protein-protein, and protein-DNA complexes. J. Med. Chem. 2005, 48, 2325–2335. [Google Scholar] [CrossRef]
- Muegge, I.; Martin, Y.C. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J. Med. Chem. 1999, 42, 791–804. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Mikami, Y.; Kubota, S.; Nakamura, H. Multiple target screening method for robust and accurate in silico ligand screening. J. Mol. Graphics Modell. 2005, 25, 61–70. [Google Scholar]
- Shan, Y.; Kim, T.E.; Eastwood, M.P.; Dror, R.O.; Seeliger, M.A.; Shaw, D.E. How does a drug molecule find its target binding site? J. Am. Chem. Soc. 2011, 133, 9181–9183. [Google Scholar] [CrossRef]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Soc. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef]
- Kamiya, N.; Yonezawa, Y.; Nakamura, H.; Higo, J. Protein-inhibitor flexible docking by a multicanonical sampling: Native complex structure with the lowest free energy and a free-energy barrier distinguishing the native complex from the others. Proteins 2008, 70, 41–53. [Google Scholar]
- Nakajima, N; Higo, J; Kidera, A; Nakamura, H. Flexible docking of a ligand peptide to a receptor protein by multicanonical molecular dynamics simulation. Chem. Phys. Lett. 1997, 278, 297–301. [Google Scholar] [CrossRef]
- Berg, B.A.; Neuhaus, T. Multicanonical algorithms for first order phase transitions. Phys. Lett. B 1991, 267, 249–253. [Google Scholar] [CrossRef]
- Nakajima, N.; Nakamura, H.; Kidera, A. Multicanonical ensemble generated by molecular dynamics simulation for enhanced conformational sampling of peptides. J. Phys. Chem. B 1997, 101, 817–824. [Google Scholar]
- Kim, J.G.; Fukunishi, Y.; Nakamura, H. Multicanonical molecular dynamics algorithm employing adaptive force-biased iteration scheme. Phys. Rev. E 2004. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Mikami, Y.; Nakamura, H. The filling potential method: A method for estimating the free energy surface for protein-ligand docking. J. Phys. Chem. B 2003, 107, 13201–13210. [Google Scholar] [CrossRef]
- Gervasio, F.L.; Laio, A.; Parrinello, M. Flexible docking in solution using metadynamics. J. Am. Chem. Soc. 2005, 127, 2600–2607. [Google Scholar] [CrossRef]
- Branduardi, D.; Gervasio, F.L.; Parrinello, M. From A to B in free energy space. J. Chem. Phys. 2007, 054103. [Google Scholar]
- Fukunishi, Y.; Mitomo, D.; Nakamura, H. Protein-ligand binding free energy calculation by the smooth reaction path generation (SRPG) method. J. Chem. Inf. Model. 2009, 49, 1944–1951. [Google Scholar] [CrossRef]
- Liphardt, J.; Dumont, S.; Smith, S.B.; Tinoco I., Jr.; Bustamante, C. Equilibrium information from nonequilibrium measurements in an experimental test of Jarzynski’s equality. Science 2002, 296, 1832–1835. [Google Scholar] [CrossRef]
- Fujitani, H.; Tanida, Y.; Matsuura, A. Massively parallel computation of absolute binding free energy with well-equilibrated states. Phys. Rev. E 2009, 021914. [Google Scholar]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput-Aided. Mol. Des. 1998, 12, 27–35. [Google Scholar]
- Ortiz, A.R.; Pisabarro, M.T.; Gago, F.; Wade, R.C. Prediction of drug binding affinities by comparative binding energy analysis. J. Med. Chem. 1995, 38, 2681–2691. [Google Scholar] [CrossRef]
- Cuevas, C.; Pastor, M.; Perez, C.; Gago, F. Comparative binding energy (COMBINE) analysis of human neutrophil elastase inhibition by pyridone-containing trifluoromethylketones. Comb. Chem. High Throughput Screen. 2001, 4, 627–642. [Google Scholar]
- Perez, C.; Pastor, M.; Ortiz, A.R.; Gago, F. Comparative binding energy analysis of HIV-1 protease inhibitors: incorporation of solvent effects and validation as a powerful tool in receptor-based drug design. J. Med. Chem. 1998, 41, 836–852. [Google Scholar] [CrossRef]
- Lozano, J.J.; Pastor, M.; Cruciani, G.; Gaedt, K.; Centeno, N.B.; Gago, F.; Sanz, F. 3D-QSAR methods on the basis of ligand-receptor complexes. Application of COMBINE and GRID/GOLPE methodologies to a series of CYP1A2 ligands. J. Comput. Aided Mol. Des. 2000, 14, 341–353. [Google Scholar]
- Tomic, S.; Nilsson, L.; Wade, R.C. Nuclear receptor-DNA binding specificity: A COMBINE and Free-Wilson QSAR analysis. J. Med. Chem. 2000, 43, 1780–1792. [Google Scholar] [CrossRef]
- Wang, T.; Wade, R.C. Comparative binding energy (COMBINE) analysis of influenza neuraminidase-inhibitor complexes. J. Med. Chem. 2001, 44, 961–971. [Google Scholar] [CrossRef]
- Murcia, M.; Ortiz, A.R. Virtual screening with flexible docking and COMBINE-based models. Application to a series of factor Xa inhibitors. J. Med. Chem. 2004, 47, 805–820. [Google Scholar] [CrossRef]
- Zwanzig, R.W. High-temperature equation of state by a perturbation method. I. Nonpolar gases. J. Chem. Phys. 1954, 22, 1420–1426. [Google Scholar] [CrossRef]
- Kubo, R. Generalized cumulant expansion method. J. Phys. Soc. Jpn. 1962, 17, 1100–1120. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E. III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Merz, K.M.; Wang, B.; Pearlman, D.A.; et al. AMBER 8; University of California: San Francisco, CA, USA, 2004. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Compt. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Hawkins, D.G.; Cramer, J.C.; Truhlar, G.D. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Watanabe, Y.S.; Kim, J.; Fukunishi, Y.; Nakamura, H. Free energy Landscape of small peptides in an implicit solvent model determined by force-biased multicanonical dynamics simulation. Chem. Phys. Letts. 2004, 400, 258–263. [Google Scholar] [CrossRef]
- Lyne, P.D.; Lamb, M.L.; Saeh, J.C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 2006, 49, 4805–4808. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E., Jr.; Burant, J.C.; et al. Gaussian 98 (Revision A.9); Gaussian Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating lipid water. J Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Greengard, L.; Rokhlin, V. A fast algorithm for particle simulations. J. Comput. Phys. 1987, 73, 325–348. [Google Scholar] [CrossRef]
- MyPresto, version 4; a program suite composed of several molecular simulations for drug development; Osaka University: Osaka, Japan, 2012.
- Sample Availability: Samples of the compounds and proteins are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fukunishi, Y.; Nakamura, H. Statistical Estimation of the Protein-Ligand Binding Free Energy Based On Direct Protein-Ligand Interaction Obtained by Molecular Dynamics Simulation. Pharmaceuticals 2012, 5, 1064-1079. https://doi.org/10.3390/ph5101064
Fukunishi Y, Nakamura H. Statistical Estimation of the Protein-Ligand Binding Free Energy Based On Direct Protein-Ligand Interaction Obtained by Molecular Dynamics Simulation. Pharmaceuticals. 2012; 5(10):1064-1079. https://doi.org/10.3390/ph5101064
Chicago/Turabian StyleFukunishi, Yoshifumi, and Haruki Nakamura. 2012. "Statistical Estimation of the Protein-Ligand Binding Free Energy Based On Direct Protein-Ligand Interaction Obtained by Molecular Dynamics Simulation" Pharmaceuticals 5, no. 10: 1064-1079. https://doi.org/10.3390/ph5101064
APA StyleFukunishi, Y., & Nakamura, H. (2012). Statistical Estimation of the Protein-Ligand Binding Free Energy Based On Direct Protein-Ligand Interaction Obtained by Molecular Dynamics Simulation. Pharmaceuticals, 5(10), 1064-1079. https://doi.org/10.3390/ph5101064