Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges

Abstract
:
1. Introduction
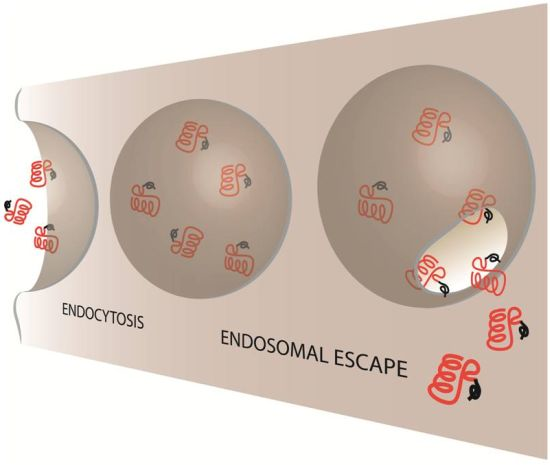
2. Evidence of CPP-Mediated Endosomal Escape

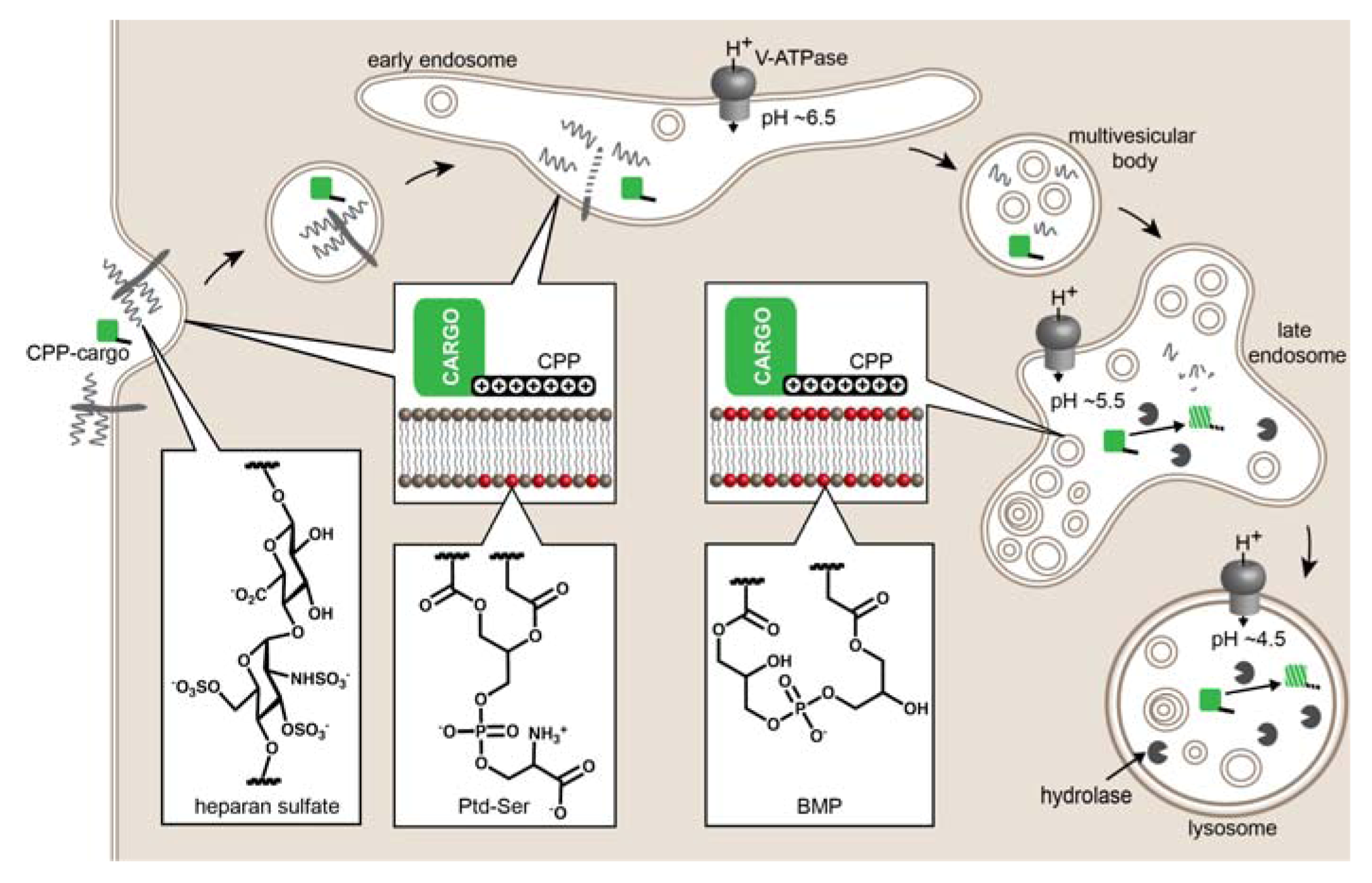
Mechanisms of Endosomal Escape
3. Strategies to Improve Endosomal Release of CPP-Cargos
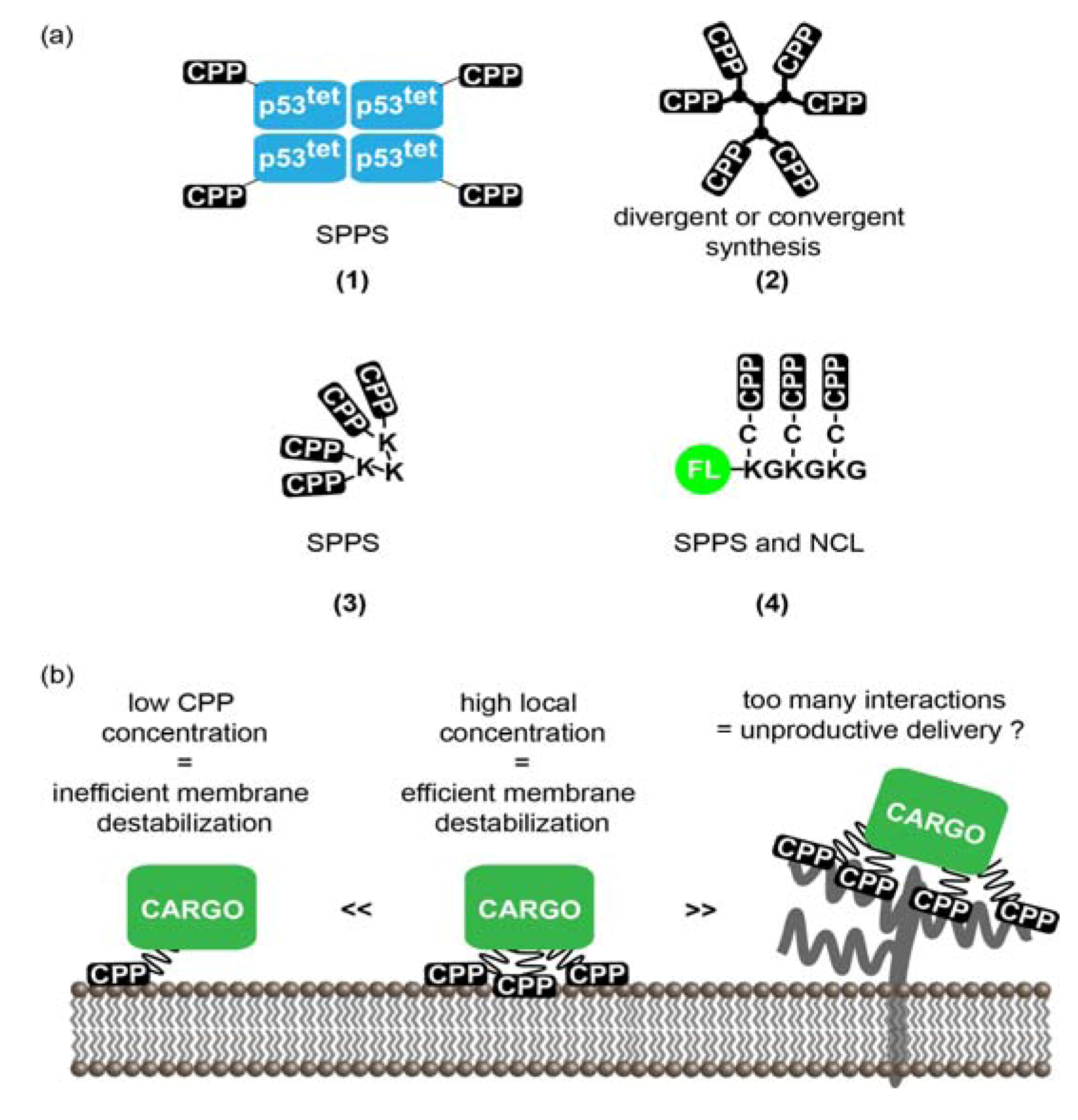
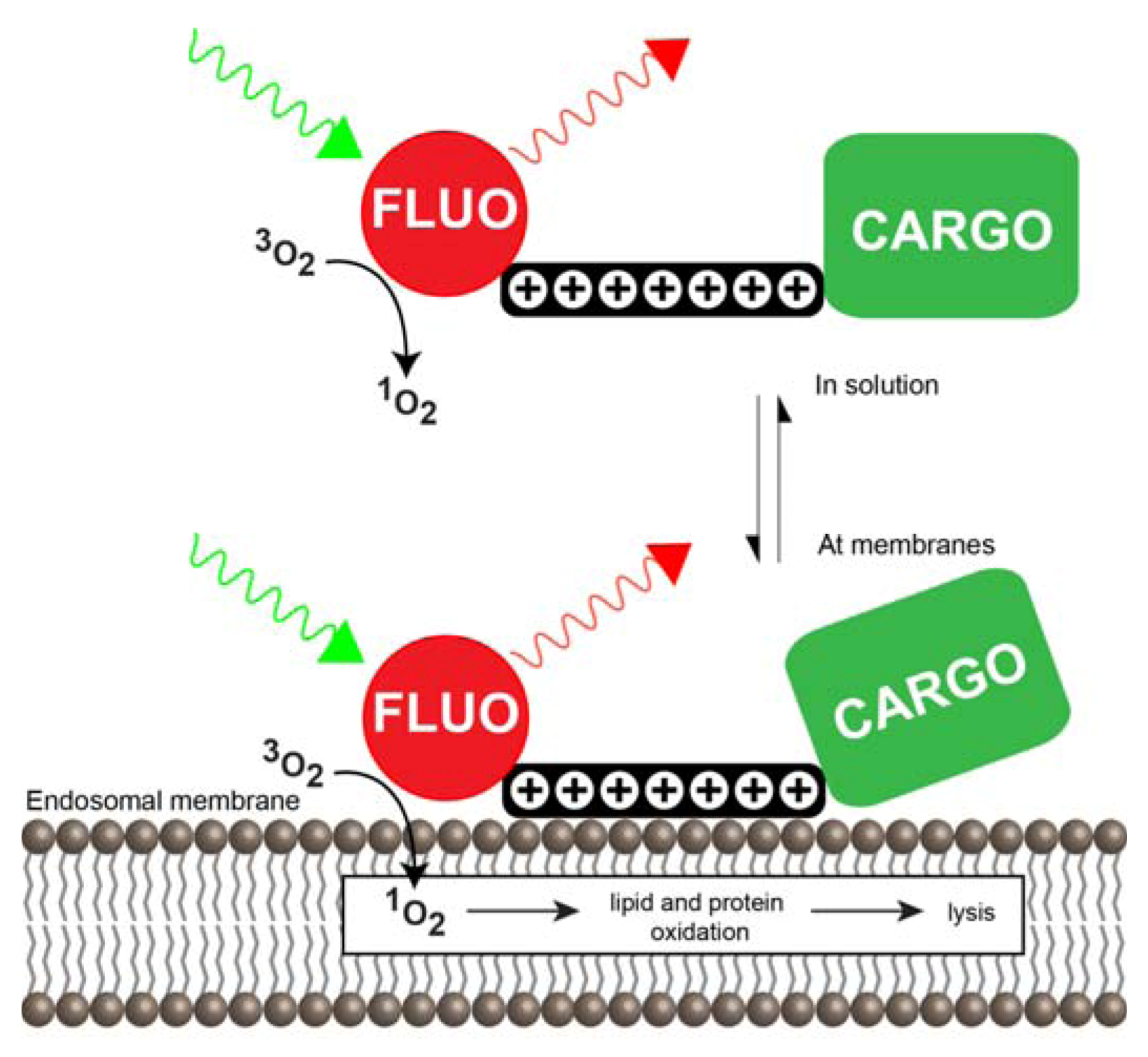
3.1. Multivalent CPPs
3.1.1. Multivalency: Concept and Rationale for the Increase of CPPs Activity
3.1.2. Strategies to Generate MCPPs

3.1.3. MCPPs Mediated Delivery
3.1.4. Limitations and Future Challenges
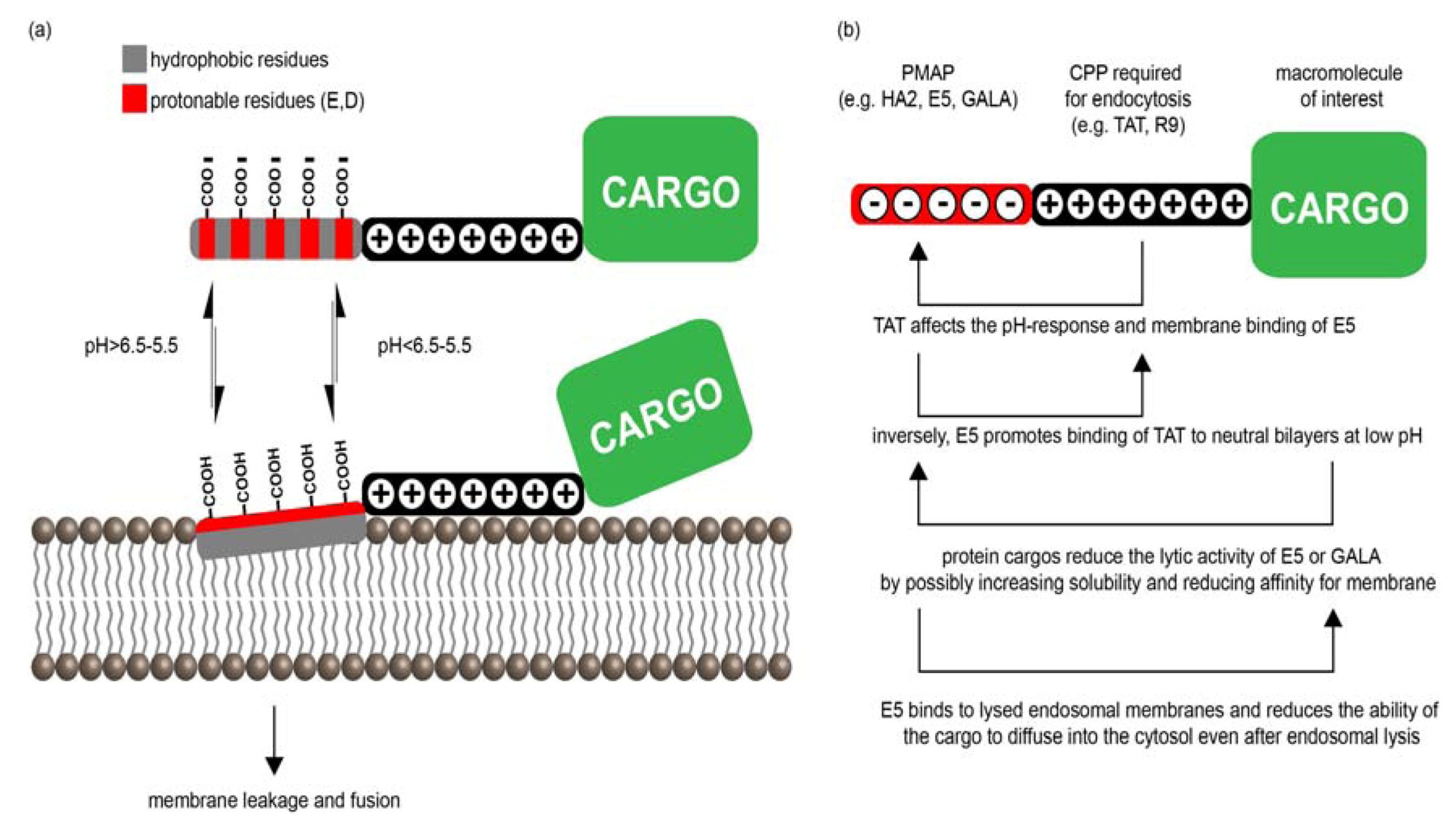
3.2. PMAPs (pH-Dependent Membrane Active Peptides)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Cargo | Ref. |
|---|---|---|---|
| HA2E5-TAT | GLFEAIAEFIENGWEGLIEGWYG | mCherry, Fluorescently labeled dextran | [121] |
| HA2-penetratin | GLFGAIAGFIENGWEGMIDGRQIKIWFQNRRMKWKK-amide | Penetratin:siRNA complex (50:1) | [122] |
| HA-K4 | GLFGAIAGFIENGWEGMIDG-SSKKKK | Plasmid DNA, plasmid DNA+ lipofectamine™ | [123] |
| GS-HA2: HA2-coated gelatin-silica nanoparticles (GSNP) | GDIMGEWGNEIFGAIAGFLGC (coating thru disulfide bond) | Plasmid DNA (pGL3) | [124] |
| GS-TH: Tat and HA2-coated GSNP (coating thru disulfide bond) | |||
| HA2E4 | GLFEAIAGFIENGWEGMIDG GGYC | EGF-poly lysine and BODIPY-labeled antisense oligonucleotide (ONs) complex | [125] |
| Biotinylated TAT-HA2 | (Biotin-CKYGRRRQRRKKRG-GDIMG EWGNE IFGAI AGFLG | Anti-biotin antibody coated gold nanoparticles | [126] |
| GALA | WEAALAEALAEALAEHLAEALAEALEALAA | siRNA, Nanoparticles | [127,128] |
| INF-7--(PEG)6-NH | GLFEAIEGFIENGWEGMIDG WYG-(PEG)6-NH2 | Fluorescently labeled TAT-NeutrAvidin | [129] |
| GALA-INF3-(PEG)6-NH | GLFEAIEGFIENGWEGLAEALAEALEALAA-(PEG)6-NH2 | Fluorescently labeled TAT-NeutrAvidin | [129] |
| GALA-INF3-(PEG)6-NH | GLFEAIEGFIENGWEGLAEALAEALEALAA-(PEG)6-NH2 | Fluorescently labeled TAT-NeutrAvidin | [129] |
| INF-7 | GLFEAIEGFIENGWEGMIDG WYG | Polyplex | [130,131,132] |
| diINF-7 | GLFEAIEGFIENGWEGMIDG WYGC (dimerizing through Cys) | siRNA, DNA, immunoliposome encapsulated diphtheria toxin A chain (DTA) | [133,134,135] |
| INF7-SGSCG | GLFEAIEGFIENGWEGMIWDYG-SGSCG | Polyplex (pCMVLuc:K8) | [136] |
| INF7-K(GalNAc)2 | GLFEAIEGFIENGWEGMIWDYG-SGSC-K(GalNAc)2 | Polyplex (pCMVLuc:K8) | [136] |
3.2.1. PMAP-CPP Chimeras
3.2.2. Mechanisms of PMAP-CPP Mediated Endosomal Escape

3.2.3. PMAP Interactions with CPPs and Cargos
3.2.4. Toxicity
3.2.5. Future Challenges
3.3. Photochemical Internalization Using CPPs
3.3.1. CPP-Mediated Photochemical Internalization
3.3.2. Mechanisms of CPP-Mediated PCI

3.3.3. Cell Death
3.3.4. Future Challenges
4. Conclusions
Conflict of Interest
Acknowledgements
References
- Brooks, H.; Lebleu, B.; Vivés, E. Tat peptide-mediated cellular delivery: back to basics. Adv. Drug Deliv. Rev. 2005, 57, 559–577. [Google Scholar] [CrossRef]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. British J. Pharm. 2009, 157, 195–206. [Google Scholar] [CrossRef]
- Nakase, I.; Takeuchi, T.; Tanaka, G.; Futaki, S. Methodological and cellular aspects that govern the internalization mechanisms of arginine-rich cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 598–607. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar]
- Pooga, M.; Hallbrink, M.; Zorko, M.; Langel, Ü. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar]
- Matsushita, M.; Tomizawa, K.; Moriwaki, A.; Li, S.-T.; Terada, H.; Matsui, H. A High-Efficiency Protein Transduction System Demonstrating the Role of PKA in Long-Lasting Long-Term Potentiation. J. Neurosci. 2001, 21, 6000–6007. [Google Scholar]
- Nishikawa, M.; Otsuki, T.; Ota, A.; Guan, X.; Takemoto, S.; Takahashi, Y.; Takakura, Y. Induction of tumor-specific Immune Response by Gene Transfer of Hsp70-cell-penetrating Peptide Fusion Protein to Tumors in Mice. Mol. Therap. 2009, 18, 421–428. [Google Scholar]
- Deshayes, S.; Morris, M.C.; Divita, G.; Heitz, F. Interactions of amphipathic CPPs with model membranes. Biochim. Biophys. Acta (Biomemb.) 2006, 1758, 328–335. [Google Scholar] [CrossRef]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotech. 2001, 19, 1173–1176. [Google Scholar] [CrossRef]
- Lorents, A.; Kodavali, P.K.; Oskolkov, N.; Langel, Ü.; Hällbrink, M.; Pooga, M. Cell-penetrating Peptides Split into Two Groups Based on Modulation of Intracellular Calcium Concentration. J. Biol. Chem. 2012, 287, 16880–16889. [Google Scholar]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.V.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. British J. Pharm. 2005, 145, 1093–1102. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar]
- Allinquant, B.; Hantraye, P.; Mailleux, P.; Moya, K.; Bouillot, C.; Prochiantz, A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J. Cell Biol. 1995, 128, 919–927. [Google Scholar] [CrossRef]
- Kato, D.; Miyazawa, K.; Ruas, M.; Starborg, M.; Wada, I.; Oka, T.; Sakai, T.; Peters, G.; Hara, E. Features of replicative senescence induced by direct addition of antennapedia-p16INK4A fusion protein to human diploid fibroblasts. FEBS Lett. 1998, 427, 203–208. [Google Scholar] [CrossRef]
- Nagahara, H.; Vocero-Akbani, A.M.; Snyder, E.L.; Ho, A.; Latham, D.G.; Lissy, N.A.; Becker-Hapak, M.; Ezhevsky, S.A.; Dowdy, S.F. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998, 4, 1449–1452. [Google Scholar] [CrossRef]
- Eguchi, A.; Dowdy, S.F. siRNA delivery using peptide transduction domains. Trends in Pharm. Sci. 2009, 30, 341–345. [Google Scholar] [CrossRef]
- Rydström, A.; Deshayes, S.; Konate, K.; Crombez, L.; Padari, K.; Boukhaddaoui, H.; Aldrian, G.; Pooga, M.; Divita, G. Direct Translocation as Major Cellular Uptake for CADY Self-Assembling Peptide-Based Nanoparticles. PLoS One 2011, 6, e25924. [Google Scholar]
- Mäger, I.; Eiríksdóttir, E.; Langel, K.; El-Andaloussi, S.; Langel, Ü. Assessing the uptake kinetics and internalization mechanisms of cell-penetrating peptides using a quenched fluorescence assay. Biochim. Biophys. Acta (Biomemb.) 2010, 1798, 338–343. [Google Scholar] [CrossRef]
- Jarver, P.; Langel, K.; El-Andaloussi, S.; Langel, Ü. Applications of cell-penetrating peptides in regulation of gene expression. Biochem. Soc. Transac. 2007, 35, 770–774. [Google Scholar] [CrossRef]
- Chao, T.-Y.; Raines, R.T. Mechanism of Ribonuclease A Endocytosis: Analogies to Cell-Penetrating Peptides. Biochemistry 2011, 50, 8374–8382. [Google Scholar] [CrossRef]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Graslund, A. Mechanism of celullar uptake of cell-penetrating peptides. J. Biophys. 2011. [Google Scholar]
- Abes, S.; Williams, D.; Prevot, P.; Thierry, A.; Gait, M.J.; Lebleu, B. Endosome trapping limits the efficiency of splicing correction by PNA-oligolysine conjugates. J. Control. Release 2006, 110, 595–604. [Google Scholar] [CrossRef]
- Jones, A.T. Macropinocytosis: searching for an endocytic identity and role in the uptake of cell penetrating peptides. J. Cell. Mol. Med. 2007, 11, 670–684. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Dowdy, S.F. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends in Pharm. Sci. 2000, 21, 45–48. [Google Scholar] [CrossRef]
- Hayashi, Y.; Yamauchi, J.; Khalil, I.A.; Kajimoto, K.; Akita, H.; Harashima, H. Cell penetrating peptide-mediated systemic siRNA delivery to the liver. Int. J. Pharm. 2011, 419, 308–313. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Datta, S.; Pellois, J.-P. Real-Time Fluorescence Detection of Protein Transduction into Live Cells. J. Am. Chem. Soc. 2008, 130, 2398–2399. [Google Scholar] [CrossRef]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating Peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar]
- Fuchs, S.M.; Raines, R.T. Pathway for Polyarginine Entry into Mammalian Cells. Biochemistry 2004, 43, 2438–2444. [Google Scholar]
- Al-Taei, S.; Penning, N.A.; Simpson, J.C.; Futaki, S.; Takeuchi, T.; Nakase, I.; Jones, A.T. Intracellular traffic and fate of protein transduction domains HIV-1 TAT peptide and octaarginine. Implications for their utilization as drug delivery vectors. Bioconjug. Chem. 2005, 17, 90–100. [Google Scholar]
- Nishi, K.; Saigo, K. Cellular Internalization of green fluorescent protein fused with herpes simplex virus protein VP22 via a lipid raft-mediated endocytic pathway independent of caveolae and rho family GTPases but dependent on dynamin and Arf6. J. Biol. Chem. 2007, 282, 27503–27517. [Google Scholar] [CrossRef]
- Gillmeister, M.P.; Betenbaugh, M.J.; Fishman, P.S. Cellular Trafficking and photochemical internalization of cell penetrating peptide linked cargo proteins: A dual fluorescent labeling study. Bioconjug. Chem. 2011, 22, 556–566. [Google Scholar]
- Turner, J.J.; Arzumanov, A.A.; Gait, M.J. Synthesis, cellular uptake and HIV-1 Tat-dependent trans-activation inhibition activity of oligonucleotide analogues disulphide-conjugated to cell-penetrating peptides. Nucl. Acids Res. 2005, 33, 27–42. [Google Scholar] [CrossRef]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef]
- Ziegler, A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv. Drug Deliv. Rev. 2008, 60, 580–597. [Google Scholar]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A Comprehensive Model for the Cellular Uptake of Cationic Cell-penetrating Peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell Internalization of the Third Helix of the Antennapedia Homeodomain Is Receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar]
- Berlose, J.-P.; Convert, O.; Derossi, D.; Brunissen, A.; Chassaing, G. Conformational and Associative Behaviours of the Third Helix of Antennapedia Homeodomain in Membrane-Mimetic Environments. Euro. J. Biochem. 1996, 242, 372–386. [Google Scholar]
- Lundberg, P.; Langel, Ü. A brief introduction to cell-penetrating peptides. J. Mol. Recognit. 2003, 16, 227–233. [Google Scholar] [CrossRef]
- Trabulo, S.; Cardoso, A.L.; Mano, M.; De Lima, M.C.P. Cell-Penetrating peptides—Mechanisms of cellular uptake and generation of delivery systems. Pharmaceuticals 2010, 3, 961–993. [Google Scholar] [CrossRef] [Green Version]
- Kosuge, M.; Takeuchi, T.; Nakase, I.; Jones, A.T.; Futaki, S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjug. Chem. 2008, 19, 656–664. [Google Scholar] [CrossRef]
- Palm-Apergi, C.; Lorents, A.; Padari, K.; Pooga, M.; Hallbrink, M. The membrane repair response masks membrane disturbances caused by cell-penetrating peptide uptake. FASEB J 2009, 23, 214–223. [Google Scholar] [CrossRef]
- Futaki, S. Oligoarginine vectors for intracellular delivery: Design and cellular-uptake mechanisms. Peptide Sci. 2006, 84, 241–249. [Google Scholar]
- Fischer, R.; Fotin-Mleczek, M.; Hufnagel, H.; Brock, R. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. ChemBioChem 2005, 6, 2126–2142. [Google Scholar] [CrossRef]
- Khalil, I.A.; Kogure, K.; Futaki, S.; Harashima, H. High Density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J. Biol. Chem. 2006, 281, 3544–3551. [Google Scholar]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102, 247–253. [Google Scholar] [CrossRef]
- Poon, G.M.; Gariépy, J. Cell-surface proteoglycans as molecular portals for cationic peptide and polymer entry into cells. Biochem. Soc. Trans. 2007, 35, 788–793. [Google Scholar] [CrossRef]
- Nakase, I.; Tadokoro, A.; Kawabata, N.; Takeuchi, T.; Katoh, H.; Hiramoto, K.; Negishi, M.; Nomizu, M.; Sugiura, Y.; Futaki, S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry 2006, 46, 492–501. [Google Scholar]
- Melikov, K.; Chernomordik, L. Arginine-rich cell penetrating peptides: from endosomal uptake to nuclear delivery. Cell. Mol. Life Sci. 2005, 62, 2739–2749. [Google Scholar] [CrossRef]
- Wender, P.A.; Galliher, W.C.; Goun, E.A.; Jones, L.R.; Pillow, T.H. The design of guanidinium-rich transporters and their internalization mechanisms. Adv. Drug Deliv. Rev. 2008, 60, 452–472. [Google Scholar] [CrossRef]
- Mishra, A.; Lai, G.H.; Schmidt, N.W.; Sun, V.Z.; Rodriguez, A.R.; Tong, R.; Tang, L.; Cheng, J.; Deming, T.J.; Kamei, D.T.; Wong, G.C.L. Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 16883–16888. [Google Scholar]
- Gump, J.M.; June, R.K.; Dowdy, S.F. Revised role of glycosaminoglycans in TAT protein transduction domain-mediated cellular transduction. J. Biol. Chem. 2010, 285, 1500–1507. [Google Scholar] [CrossRef]
- Kasim, V.; Miyagishi, M.; Taira, K. Control of siRNA expression using the Cre–loxP recombination system. Nucl. Acids Research 2004, 32, e66–e66. [Google Scholar]
- El-Andaloussi, S.; Johansson, H.J.; Lundberg, P.; Langel, Ü. Induction of splice correction by cell-penetrating peptide nucleic acids. J. Gene Med. 2006, 8, 1262–1273. [Google Scholar] [CrossRef]
- Appelbaum, J.S.; LaRochelle, J.R.; Smith, B.A.; Balkin, D.M.; Holub, J.M.; Schepartz, A. Arginine topology controls escape of minimally cationic proteins from early endosomes to the cytoplasm. Chem. Biol. 2012, 19, 819–830. [Google Scholar] [CrossRef]
- Burlina, F.; Sagan, S.; Bolbach, G.; Chassaing, G. A direct approach to quantification of the cellular uptake of cell-penetrating peptides using MALDI-TOF mass spectrometry. Nat. Protocols 2006, 1, 200–205. [Google Scholar]
- Paramelle, D.; Subra, G.; Vezenkov, L.L.; Maynadier, M.; André, C.; Enjalbal, C.; Calmès, M.; Garcia, M.; Martinez, J.; Amblard, M. A Straightforward approach for Cellular-Uptake quantification. Angewandte Chemie Int. Edition 2010, 49, 8240–8243. [Google Scholar] [CrossRef]
- Walrant, A.; Correia, I.; Jiao, C.-Y.; Lequin, O.; Bent, E.H.; Goasdoué, N.; Lacombe, C.; Chassaing, G.; Sagan, S.; Alves, I.D. Different membrane behaviour and cellular uptake of three basic arginine-rich peptides. Biochim. Biophys. Acta (Biomemb.) 2011, 1808, 382–393. [Google Scholar] [CrossRef]
- Alves, I.D.; Bechara, C.; Walrant, A.; Zaltsman, Y.; Jiao, C.-Y.; Sagan, S. Relationships between membrane binding, affinity and cell internalization efficacy of a cell-penetrating peptide: Penetratin as a case study. PLoS One 2011, 6, e24096. [Google Scholar]
- Takeuchi, T.; Kosuge, M.; Tadokoro, A.; Sugiura, Y.; Nishi, M.; Kawata, M.; Sakai, N.; Matile, S.; Futaki, S. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chem. Biol. 2006, 1, 299–303. [Google Scholar]
- Medintz, I.L.; Pons, T.; Delehanty, J.B.; Susumu, K.; Brunel, F.M.; Dawson, P.E.; Mattoussi, H. Intracellular delivery of quantum dot−protein cargos mediated by cell penetrating peptides. Bioconjug. Chem. 2008, 19, 1785–1795. [Google Scholar] [CrossRef]
- Pan, C.; Lu, B.; Chen, H.; Bishop, C. Reprogramming human fibroblasts using HIV-1 TAT recombinant proteins OCT4, SOX2, KLF4 and c-MYC. Mol. Biol. Reports 2010, 37, 2117–2124. [Google Scholar] [CrossRef]
- Loison, F.; Nizard, P.; Sourisseau, T.; Le Goff, P.; Debure, L.; Le Drean, Y.; Michel, D. A ubiquitin-based assay for the cytosolic uptake of protein transduction domains. Mol. Therap. 2005, 11, 205–214. [Google Scholar]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20805–20810. [Google Scholar] [CrossRef]
- Thorén, P.E.G.; Persson, D.; Esbjörner, E.K.; Goksör, M.; Lincoln, P.; Nordén, B. Membrane binding and translocation of cell-penetrating peptides. Biochemistry 2004, 43, 3471–3489. [Google Scholar]
- Tiriveedhi, V.; Butko, P. A fluorescence spectroscopy study on the interactions of the TAT-PTD peptide with model lipid membranes. Biochemistry 2007, 46, 3888–3895. [Google Scholar] [CrossRef]
- Leventis, P.A.; Grinstein, S. The distribution and function of phosphatidylserine in cellular membranes. Ann. Rev. Biophys. 2010, 39, 407–427. [Google Scholar] [CrossRef]
- Kay, J.G.; Grinstein, S. Sensing phosphatidylserine in cellular membranes. Sensors 2011, 11, 1744–1755. [Google Scholar]
- Ruzza, P.; Biondi, B.; Marchiani, A.; Antolini, N.; Calderan, A. Cell-penetrating peptides: A comparative study on lipid affinity and cargo delivery properties. Pharmaceuticals 2010, 3, 1045–1062. [Google Scholar] [CrossRef]
- Lee, Y.J.; Johnson, G.; Pellois, J.P. Modeling of the endosomolytic activity of HA2-TAT peptides with red blood cells and ghosts. Biochemistry 2010, 49, 7854–7866. [Google Scholar]
- Cahill, K. Molecular electroporation and the transduction of oligoarginines. Physic. Biol. 2009, 7, 1–14. [Google Scholar]
- Tünnemann, G.; Ter-Avetisyan, G.; Martin, R.M.; Stöckl, M.; Herrmann, A.; Cardoso, M.C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Peptide Sci. 2008, 14, 469–476. [Google Scholar] [CrossRef]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Ann. Rev. Biochem. 1999, 68, 729–777. [Google Scholar]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar]
- Rothe, R.; Liguori, L.; Villegas-Mendez, A.; Marques, B.; Grunwald, D.; Drouet, E.; Lenormand, J.-L. Characterization of the cell-penetrating properties of the epstein-barr virus ZEBRA trans-activator. J. Biol. Chem. 2010, 285, 20224–20233. [Google Scholar]
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2001, 276, 3254–3261. [Google Scholar]
- Belting, M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends in Biochem. Sci. 2003, 28, 145–151. [Google Scholar] [CrossRef]
- Yanagishita, M.; Hascall, V.C. Metabolism of proteoglycans in rat ovarian granulosa cell culture. Multiple intracellular degradative pathways and the effect of chloroquine. J. Biol. Chem. 1984, 259, 10270–10283. [Google Scholar]
- Magzoub, M.; Kilk, K.; Eriksson, L.E.G.; Langel, Ü.; Graslund, A. Interaction and structure induction of cell-penetrating peptides in the presence of phospholipid vesicles. Biochim. Biophys. Acta (Biomemb.) 2001, 1512, 77–89. [Google Scholar] [CrossRef]
- Kobayashi, T.; Startchev, K.; Whitney, A.J.; Gruenberg, J. Localization of lysobisphosphatidic acid-rich memmbrane domains in late endosomes. Biol. Chem. 2001, 382, 483–485. [Google Scholar]
- Matsuo, H.; Chevallier, J.; Mayran, N.; Le Blanc, I.; Ferguson, C.; Faure, J.; Blanc, N.S.; Matile, S.; Dubochet, J.; Sadoul, R.m.; Parton, R.G.; Vilbois, F.; Gruenberg, J. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 2004, 303, 531–534. [Google Scholar]
- Yang, S.-T.; Zaitseva, E.; Chernomordik, L.V.; Melikov, K. Cell-penetrating peptide induces leaky fusion of liposomes containing late endosome-specific anionic lipid. Biophys. J. 2010, 99, 2525–2533. [Google Scholar] [CrossRef]
- Kawamura, K.S.; Sung, M.; Bolewska-Pedyczak, E.; Gariépy, J. Probing the impact of valency on the routing of arginine-rich peptides into eukaryotic cells. Biochemistry 2006, 45, 1116–1127. [Google Scholar]
- Hassane, F.S.; Ivanova, G.D.; Bolewska-Pedyczak, E.; Abes, R.; Arzumanov, A.A.; Gait, M.J.; Lebleu, B.; Gariépy, J. A peptide-based dendrimer that enhances the splice-redirecting activity of PNA Conjugates in cells. Bioconjug. Chem. 2009, 20, 1523–1530. [Google Scholar] [CrossRef]
- Kang, H.; DeLong, R.; Fisher, M.; Juliano, R. Tat-conjugated PAMAM dendrimers as delivery agents for antisense and siRNA oligonucleotides. Pharm. Res. 2005, 22, 2099–2106. [Google Scholar]
- Juliano, R.L. Intracellular delivery of oligonucleotide conjugates and dendrimer complexes. Annals NY Acad. Sci. 2006, 1082, 18–26. [Google Scholar] [CrossRef]
- Pantos, A.; Tsiourvas, D.; Nounesis, G.; Paleos, C.M. Interaction of functional dendrimers with multilamellar liposomes:Design of a model system for studying drug delivery. Langmuir 2005, 21, 7483–7490. [Google Scholar] [CrossRef]
- Kim, J.-B.; Choi, J.S.; Nam, K.; Lee, M.; Park, J.-S.; Lee, J.-K. Enhanced transfection of primary cortical cultures using arginine-grafted PAMAM dendrimer, PAMAM-Arg. J. Control. Release 2006, 114, 110–117. [Google Scholar] [CrossRef]
- Medina, S.H.; El-Sayed, E.H. Dendrimers as carriers for delivery of chemotherapeutic agents. Chem. Rev. 2009, 109, 3141–3157. [Google Scholar] [CrossRef]
- Sheldon, K.; Liu, D.; Ferguson, J.; Gariépy, J. Loligomers: Design of de novo peptide-based intracellular vehicles. Proc. Natl. Acad. Sci. USA 1995, 92, 2056–2060. [Google Scholar] [CrossRef]
- Singh, D.; Kiarash, R.; Kawamura, K.; LaCasse, E.C.; Gariépy, J. Penetration and intracellular routing of nucleus-directed peptide-based shuttles (Loligomers) in eukaryotic cells. Biochemistry 1998, 37, 5798–5809. [Google Scholar] [CrossRef]
- Kawamura, K.S.; Su, R.-C.; Nguyen, L.T.; Elford, A.R.; Ohashi, P.S.; Gariépy, J. In vivo generation of cytotoxic t cells from epitopes displayed on peptide-based delivery vehicles. J. Immun. 2002, 168, 5709–5715. [Google Scholar]
- Singh, D.; Bisland, S.K.; Kawamura, K.; Gariépy, J. Peptide-based intracellular shuttle able to facilitate gene transfer in mammalian cells. Bioconjug. Chem. 1999, 10, 745–754. [Google Scholar] [CrossRef]
- Angeles-Boza, A.M.; Erazo-Oliveras, A.; Lee, Y.-J.; Pellois, J.-P. Generation of endosomolytic reagents by branching of cell-penetrating peptides: tools for the delivery of bioactive compounds to live cells in cis or trans. Bioconjug. Chem. 2010, 21, 2164–2167. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar]
- Rudolph, C.; Schillinger, U.; Ortiz, A.; Tabatt, K.; Plank, C.; Müller, R.H.; Rosenecker, J. Application of novel solid lipid nanoparticle (SLN)-Gene vector formulations based on a dimeric HIV-1 TAT-Peptide in vitro and in vivo. Pharm. Res. 2004, 21, 1662–1669. [Google Scholar] [CrossRef]
- Chugh, A.; Amundsen, E.; Eudes, F. Translocation of cell-penetrating peptides and delivery of their cargoes in triticale microspores. Plant Cell Reports 2009, 28, 801–810. [Google Scholar] [CrossRef]
- Lee, S.-J.; Yoon, S.-H.; Doh, K.-O. Enhancement of gene delivery using novel homodimeric tat peptide formed by disulfide bond. J. Microbiol. Biotech. 2011, 21, 802–807. [Google Scholar] [CrossRef]
- Muir, T.W.; Sondhi, D.; Cole, P.A. Expressed protein ligation: A general method for protein engineering. Proc. Natl. Acad. Sci. USA 1998, 95, 6705–6710. [Google Scholar] [CrossRef]
- Muir, T.W. Semisynthesis of proteins by expressed protein ligation. Ann. Rev. Biochem. 2003, 72, 249–289. [Google Scholar] [CrossRef]
- Sung, M.; Poon, G.; Gariepy, J. The importance of valency in enhancing the import and cell routing potential of protein transduction domain-containing molecules. Biochim. Biophys. Acta (Biomemb.) 2006, 1758, 355–363. [Google Scholar] [CrossRef]
- Tung, C.-H.; Mueller, S.; Weissleder, R. Novel branching membrane translocational peptide as gene delivery vector. Bioorg. Med. Chem. 2002, 10, 3609–3614. [Google Scholar] [CrossRef]
- Rudolph, C.; Plank, C.; Lausier, J.; Schillinger, U.; Müller, R.H.; Rosenecker, J. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J. Biol. Chem. 2003, 278, 11411–11418. [Google Scholar]
- Tam, J.P. Synthetic peptide vaccine design: synthesis and properties of a high-density multiple antigenic peptide system. Proc. Natl. Acad. Sci. USA 1988, 85, 5409–5413. [Google Scholar] [CrossRef]
- Fujita, Y.; Taguchi, H. Current status of multiple antigen-presenting peptide vaccine systems: Application of organic and inorganic nanoparticles. Chem. Central J. 2011, 5, 48. [Google Scholar] [CrossRef]
- Schmid, S.; Fuchs, R.; Kielian, M.; Helenius, A.; Mellman, I. Acidification of endosome subpopulations in wild-type Chinese hamster ovary cells and temperature-sensitive acidification-defective mutants. J. Cell Biol. 1989, 108, 1291–1300. [Google Scholar]
- Serresi, M.; Bizzarri, R.; Cardarelli, F.; Beltram, F. Real-time measurement of endosomal acidification by a novel genetically encoded biosensor. Anal. Bioanal. Chem. 2009, 393, 1123–1133. [Google Scholar] [CrossRef]
- Wharton, S.A.; Martin, S.R.; Ruigrok, R.W.; Skehel, J.J.; Wiley, D.C. Membrane fusion by peptide analogues of influenza virus haemagglutinin. J. Gen. Virol. 1988, 69, 1847–1857. [Google Scholar] [CrossRef]
- Smith, A.E.; Helenius, A. How viruses enter animal cells. Science 2004, 304, 237–242. [Google Scholar] [CrossRef]
- Han, X.; Bushweller, J.H.; Cafiso, D.S.; Tamm, L.K. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza. Nat. Struct. Biol. 2001, 8, 715–720. [Google Scholar]
- Lorieau, J.L.; Louis, J.M.; Bax, A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc. Natl. Acad. Sci. USA 2010, 107, 11341–11346. [Google Scholar] [CrossRef]
- Durrer, P.; Galli, C.; Hoenke, S.; Corti, C.; Gluck, R.; Vorherr, T.; Brunner, J. H+-induced membrane insertion of influenza virus hemagglutinin involves the HA2 amino-terminal fusion peptide but not the coiled coil region. J. Biol. Chem. 1996, 271, 13417–13421. [Google Scholar]
- Kozlov, M.M.; Chernomordik, L.V. A mechanism of protein-mediated fusion: Coupling between refolding of the influenza hemagglutinin and lipid rearrangements. Biophys. J. 1998, 75, 1384–1396. [Google Scholar] [CrossRef]
- Tamm, L.K.; Han, X. Viral fusion peptides: a tool set to disrupt and connect biological membranes. Biosci. Reports 2000, 20, 501–518. [Google Scholar]
- Esbjörner, E.K.; Oglecka, K.; Lincoln, P.; Gräslund, A.; Nordén, B. Membrane binding of pH-Sensitive influenza fusion peptides. Positioning, configuration, and induced leakage in a lipid vesicle model. Biochemistry 2007, 46, 13490–13504. [Google Scholar] [CrossRef]
- Korte, T.; Ludwig, K.; Herrmann, A. ph-dependent hydrophobicity profile of hemagglutinin of influenza virus and its possible relevance in virus fusion. Biosci. Reports 1992, 12, 397–406. [Google Scholar]
- Zhelev, D.V.; Stoicheva, N.; Scherrer, P.; Needham, D. Interaction of synthetic HA2 influenza fusion peptide analog with model membranes. Biophys. J. 2001, 81, 285–304. [Google Scholar] [CrossRef]
- Turk, M.J.; Reddy, J.A.; Chmielewski, J.A.; Low, P.S. Characterization of a novel pH-sensitive peptide that enhances drug release from folate-targeted liposomes at endosomal pHs. Biochim. Biophys, Acta (Biomemb.) 2002, 1559, 56–68. [Google Scholar] [CrossRef]
- Li, W.J.; Nicol, F.; Szoka, F.C. GALA: a designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 967–985. [Google Scholar] [CrossRef]
- Lee, Y.J.; Johnson, G.; Peltier, G.C.; Pellois, J.P. A HA2-Fusion tag limits the endosomal release of its protein cargo despite causing endosomal lysis. Biochim. Biophys, Acta (Gen. Sub.) 2011, 1810, 752–758. [Google Scholar] [CrossRef]
- Lundberg, P.; El-Andaloussi, S.; Sutlu, T.; Johansson, H.; Langel, Ü. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef]
- Subramanian, A.; Ma, H.; Dahl, K.N.; Zhu, J.; Diamond, S.L. Adenovirus or HA-2 fusogenic peptide-assisted lipofection increases cytoplasmic levels of plasmid in nondividing endothelium with little enhancement of transgene expression. J. Gene Med. 2002, 4, 75–83. [Google Scholar] [CrossRef]
- Ye, S.F.; Tian, M.M.; Wang, T.X.; Ren, L.; Wang, D.; Shen, L.H.; Shang, T. Synergistic effects of cell-penetrating peptide Tat and fusogenic peptide HA2-enhanced cellular internalization and gene transduction of organosilica nanoparticles. Nanomedicine 2012, 833–841. [Google Scholar]
- Deshpande, D.; Toledo-Velasquez, D.; Thakkar, D.; Liang, W.; Rojanasakul, Y. Enhanced cellular uptake of oligonucleotides by EGF receptor-mediated endocytosis in A549 cells. Pharm. Res. 1996, 13, 57–61. [Google Scholar] [CrossRef]
- Kumar, S.; Harrison, N.; Richards-Kortum, R.; Sokolov, K. Plasmonic nanosensors for imaging intracellular biomarkers in live cells. Nano Lett. 2007, 7, 1338–1343. [Google Scholar]
- Hatakeyama, H.; Ito, E.; Akita, H.; Oishi, M.; Nagasaki, Y.; Futaki, S.; Harashima, H. A pH-sensitive fusogenic peptide facilitates endosomal escape and greatly enhances the gene silencing of siRNA-containing nanoparticles in vitro and in vivo. J. Control. Release 2009, 139, 127–132. [Google Scholar] [CrossRef]
- Akita, H.; Masuda, T.; Nishio, T.; Niikura, K.; Ijiro, K.; Harashima, H. Improving in vivo hepatic transfection activity by controlling intracellular trafficking: The function of GALA and maltotriose. Mol. Pharm. 2011, 8, 1436–1442. [Google Scholar] [CrossRef]
- Chen, A.K.; Behlke, M.A.; Tsourkas, A. Efficient cytosolic delivery of molecular beacon conjugates and flow cytometric analysis of target RNA. Nucl. Acids Res. 2008, 36, e69. [Google Scholar]
- Funhoff, A.M.; van Nostrum, C.F.; Koning, G.A.; Schuurmans-Nieuwenbroek, N.M.; Crommelin, D.J.; Hennink, W.E. Endosomal escape of polymeric gene delivery complexes is not always enhanced by polymers buffering at low pH. Biomacromolecules 2004, 5, 32–39. [Google Scholar] [CrossRef]
- Funhoff, A.M.; van Nostrum, C.F.; Lok, M.C.; Fretz, M.M.; Crommelin, D.J.; Hennink, W.E. Poly(3-guanidinopropyl methacrylate): A novel cationic polymer for gene delivery. Bioconjug. Chem. 2004, 15, 1212–1220. [Google Scholar] [CrossRef]
- Funhoff, A.M.; van Nostrum, C.F.; Janssen, A.P.; Fens, M.H.; Crommelin, D.J.; Hennink, W.E. Polymer side-chain degradation as a tool to control the destabilization of polyplexes. Pharm. Res. 2004, 21, 170–176. [Google Scholar] [CrossRef]
- Oliveira, S.; van Rooy, I.; Kranenburg, O.; Storm, G.; Schiffelers, R.M. Fusogenic peptides enhance endosomal escape improving siRNA-induced silencing of oncogenes. Int. J. Pharm. 2007, 331, 211–214. [Google Scholar]
- Funhoff, A.M.; van Nostrum, C.F.; Lok, M.C.; Kruijtzer, J.A.; Crommelin, D.J.; Hennink, W.E. Cationic polymethacrylates with covalently linked membrane destabilizing peptides as gene delivery vectors. J. Control. Release 2005, 101, 233–246. [Google Scholar] [CrossRef]
- Mastrobattista, E.; Koning, G.A.; van Bloois, L.; Filipe, A.C.; Jiskoot, W.; Storm, G. Functional characterization of an endosome-disruptive peptide and its application in cytosolic delivery of immunoliposome-entrapped proteins. J. Biol. Chem. 2002, 277, 27135–27143. [Google Scholar]
- Van Rossenberg, S.M.; Sliedregt-Bol, K.M.; Meeuwenoord, N.J.; Van Berkel, T.J.; Van Boom, J.H.; Van Der Marel, G.A.; Biessen, E.A. Targeted lysosome disruptive elements for improvement of parenchymal liver cell-specific gene delivery. J. Biol. Chem. 2002, 277, 45803–45810. [Google Scholar]
- Lo, S.L.; Wang, S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials 2008, 29, 2408–2414. [Google Scholar] [CrossRef]
- Chang, K.L.; Higuchi, Y.; Kawakami, S.; Yamashita, F.; Hashida, M. Efficient Gene Transfection by Histidine-Modified Chitosan through Enhancement of Endosomal Escape. Bioconjug. Chem. 2010, 21, 1087–1095. [Google Scholar] [CrossRef]
- Benns, J.M.; Choi, J.S.; Mahato, R.I.; Park, J.S.; Kim, S.W. pH-sensitive cationic polymer gene delivery vehicle: N-Ac-poly(L-histidine)-graft-poly(L-lysine) comb shaped polymer. Bioconjug. Chem. 2000, 11, 637–645. [Google Scholar] [CrossRef]
- Midoux, P.; Kichler, A.; Boutin, V.; Maurizot, J.C.; Monsigny, M. Membrane permeabilization and efficient gene transfer by a peptide containing several histidines. Bioconjug. Chem. 1998, 9, 260–267. [Google Scholar] [CrossRef]
- Midoux, P.; LeCam, E.; Coulaud, D.; Delain, E.; Pichon, C. Histidine containing peptides and polypeptides as nucleic acid vectors. Somat. Cell Mol. Genet. 2002, 27, 27–47. [Google Scholar]
- Summerton, J.E. Endo-Porter: A novel reagent for safe, effective delivery of substances into cells. Ann. N. Y. Acad. Sci. 2005, 1058, 62–75. [Google Scholar] [CrossRef]
- Mutyam, V.; Puccetti, M.V.; Frisbie, J.; Goldstein, D.L.; Krane, C.M. Endo-Porter-mediated delivery of phosphorodiamidate morpholino oligos (PMOs) in erythrocyte suspension cultures from Cope's gray treefrog Hyla chrysoscelis. Biotechniques 2011, 50, 329–332. [Google Scholar]
- Nikopoulos, G.N.; Adams, T.L.; Adams, D.; Oxburgh, L.; Prudovsky, I.; Verdi, J.M. The use of Endo-Porter to deliver morpholinos in kidney organ culture. Biotechniques 2008, 44, 547–549. [Google Scholar] [CrossRef]
- Raghuraman, H.; Chattopadhyay, A. Melittin: a membrane-active peptide with diverse functions. Biosci. Reports 2007, 27, 189–223. [Google Scholar] [CrossRef]
- Pratt, J.P.; Ravnic, D.J.; Huss, H.T.; Jiang, X.Q.; Orozco, B.S.; Mentzer, S.J. Melittin-induced membrane permeability: A nonosmotic mechanism of cell death. In Vitro Cell Dev. Ann. 2005, 41, 349–355. [Google Scholar]
- Meyer, M.; Zintchenko, A.; Ogris, M.; Wagner, E. A dimethylmaleic acid-melittin-polylysine conjugate with reduced toxicity, pH-triggered endosomolytic activity and enhanced gene transfer potential. J. Gene Med. 2007, 9, 797–805. [Google Scholar]
- Meyer, M.; Philipp, A.; Oskuee, R.; Schmidt, C.; Wagner, E. Breathing life into polycations: functionalization with pH-responsive endosomolytic peptides and polyethylene glycol enables siRNA delivery. J. Am. Chem. Soc. 2008, 130, 3272–3273. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Dohmen, C.; Philipp, A.; Kiener, D.; Maiwald, G.; Scheu, C.; Ogris, M.; Wagner, E. Synthesis and biological evaluation of a bioresponsive and endosomolytic siRNA-polymer conjugate. Mol. Pharm. 2009, 6, 752–762. [Google Scholar]
- Rozema, D.B.; Ekena, K.; Lewis, D.L.; Loomis, A.G.; Wolff, J.A. Endosomolysis by masking of a membrane-active agent (EMMA) for cytoplasmic release of macromolecules. Bioconjug. Chem. 2003, 14, 51–57. [Google Scholar] [CrossRef]
- Salomone, F.; Cardarelli, F.; Di Luca, M.; Boccardi, C.; Nifosi, R.; Bardi, G.; Di Bari, L.; Serresi, M.; Beltram, F. A novel chimeric cell-penetrating peptide with membrane-disruptive properties for efficient endosomal escape. J. Control. Release 2012, 163, 293–303. [Google Scholar] [CrossRef]
- Ellerby, H.M.; Arap, W.; Ellerby, L.M.; Kain, R.; Andrusiak, R.; Rio, G.D.; Krajewski, S.; Lombardo, C.R.; Rao, R.; Ruoslahti, E.; Bredesen, D.E.; Pasqualini, R. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999, 5, 1032–1038. [Google Scholar] [CrossRef]
- Diaz-Achirica, P.; Prieto, S.; Ubach, J.; Andreu, D.; Rial, E.; Rivas, L. Permeabilization of the mitochondrial inner membrane by short cecropin-A-melittin hybrid peptides. Eur. J. Biochem. 1994, 224, 257–263. [Google Scholar] [CrossRef]
- Steinhauer, D.A.; Wharton, S.A.; Skehel, J.J.; Wiley, D.C. Studies of the membrane fusion activities of fusion peptide mutants of influenza virus hemagglutinin. J. Virol. 1995, 69, 6643–6651. [Google Scholar]
- Qiao, H.; Armstrong, R.T.; Melikyan, G.B.; Cohen, F.S.; White, J.M. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol. Biol. Cell 1999, 10, 2759–2769. [Google Scholar]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar]
- Koshman, Y.E.; Waters, S.B.; Walker, L.A.; Los, T.; de Tombe, P.; Goldspink, P.H.; Russell, B. Delivery and visualization of proteins conjugated to quantum dots in cardiac myocytes. J. Mol. Cell Cardiol. 2008, 45, 853–856. [Google Scholar] [CrossRef]
- Michiue, H.; Tomizawa, K.; Wei, F.Y.; Matsushita, M.; Lu, Y.F.; Ichikawa, T.; Tamiya, T.; Date, I.; Matsui, H. The NH2 terminus of influenza virus hemagglutinin-2 subunit peptides enhances the antitumor potency of polyarginine-mediated p53 protein transduction. J. Biol. Chem. 2005, 280, 8285–8289. [Google Scholar]
- Lee, Y.J.; Erazo-Oliveras, A.; Pellois, J.P. Delivery of macromolecules into live cells by simple co-incubation with a peptide. ChemBioChem 2010, 11, 325–330. [Google Scholar]
- Lieber, M.R.; Steck, T.L. A description of the holes in human erythrocyte membrane ghosts. J. Biol. Chem. 1982, 257, 11651–11659. [Google Scholar]
- Ciobanasu, C.; Siebrasse, J.P.; Kubitscheck, U. Cell-Penetrating HIV1 TAT Peptides Can Generate Pores in Model Membranes. Biophys. J. 2010, 99, 153–162. [Google Scholar]
- Han, X.; Tamm, L.K. pH-dependent self-association of influenza hemagglutinin fusion peptides in lipid bilayers. J. Mol. Biol. 2000, 304, 953–965. [Google Scholar] [CrossRef]
- Han, X.; Tamm, L.K. A host-guest system to study structure-function relationships of membrane fusion peptides. Proc. Natl. Acad. Sci. USA 2000, 97, 13097–13102. [Google Scholar] [CrossRef]
- Kuehne, J.; Murphy, R.M. Synthesis and characterization of membrane-active GALA-OKT9 conjugates. Bioconjug. Chem. 2001, 12, 742–749. [Google Scholar] [CrossRef]
- Hakansson, S.; Jacobs, A.; Caffrey, M. Heparin binding by the HIV-1 tat protein transduction domain. Protein Sci. 2001, 10, 2138–2139. [Google Scholar] [CrossRef]
- Hakansson, S.; Caffrey, M. Structural and dynamic properties of the HIV-1 tat transduction domain in the free and heparin-bound states. Biochemistry 2003, 42, 8999–9006. [Google Scholar] [CrossRef]
- Berg, K.; Selbo, P.K.; Prasmickaite, L.; Tjelle, T.E.; Sandvig, K.; Moan, J.; Gaudernack, G.; Fodstad, O.; Kjolsrud, S.; Anholt, H.; Rodal, G.H.; Rodal, S.K.; Hogset, A. Photochemical internalization: A novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999, 59, 1180–1183. [Google Scholar]
- Berg, K.; Weyergang, A.; Prasmickaite, L.; Bonsted, A.; Hogset, A.; Strand, M.T.; Wagner, E.; Selbo, P.K. Photochemical internalization (PCI): A technology for drug delivery. Methods Mol. Biol. 2010, 635, 133–145. [Google Scholar] [CrossRef]
- Berg, K.; Berstad, M.; Prasmickaite, L.; Weyergang, A.; Selbo, P.K.; Hedfors, I.; Hogset, A. Photochemical internalization: a new tool for gene and oligonucleotide delivery. Top Curr. Chem. 2010, 296, 251–281. [Google Scholar]
- Berg, K.; Nordstrand, S.; Selbo, P.K.; Tran, D.T.; Angell-Petersen, E.; Hogset, A. Disulfonated tetraphenyl chlorin (TPCS2a), a novel photosensitizer developed for clinical utilization of photochemical internalization. Photochem. Photobiol. Sci. 2011, 10, 1637–1651. [Google Scholar] [CrossRef]
- Selbo, P.K.; Hogset, A.; Prasmickaite, L.; Berg, K. Photochemical internalisation: A novel drug delivery system. Tumour Biol. 2002, 23, 103–112. [Google Scholar] [CrossRef]
- Selbo, P.K.; Weyergang, A.; Bonsted, A.; Bown, S.G.; Berg, K. Photochemical internalization of therapeutic macromolecular agents: A novel strategy to kill multidrug-resistant cancer cells. J. Pharm. Exp. Therap. 2006, 319, 604–612. [Google Scholar] [CrossRef]
- Mathews, M.S.; Blickenstaff, J.W.; Shih, E.C.; Zamora, G.; Vo, V.; Sun, C.H.; Hirschberg, H.; Madsen, S.J. Photochemical internalization of bleomycin for glioma treatment. J. Biomed. Opt. 2012, 17, 058001. [Google Scholar] [CrossRef]
- Maiolo, J.R., III; Ottinger, E.A.; Ferrer, M. Specific redistribution of cell-penetrating peptides from endosomes to the cytoplasm and nucleus upon laser illumination. J. Am. Chem. Soc. 2004, 126, 15376–15377. [Google Scholar] [CrossRef]
- Matsushita, M.; Noguchi, H.; Lu, Y.F.; Tomizawa, K.; Michiue, H.; Li, S.T.; Hirose, K.; Bonner-Weir, S.; Matsui, H. Photo-acceleration of protein release from endosome in the protein transduction system. FEBS Lett. 2004, 572, 221–226. [Google Scholar] [CrossRef]
- Endoh, T.; Sisido, M.; Ohtsuki, T. Spatial regulation of specific gene expression through photoactivation of RNAi. J. Control. Release 2009, 137, 241–245. [Google Scholar] [CrossRef]
- Zhao, J.F.; Chen, J.Y.; Mi, L.; Wang, P.N.; Peng, Q. Enhancement of intracellular delivery of anti-cancer drugs by the Tat peptide. Ultrastruct. Pathol. 2011, 35, 119–123. [Google Scholar] [CrossRef]
- Choi, Y.; McCarthy, J.R.; Weissleder, R.; Tung, C.H. Conjugation of a photosensitizer to an oligoarginine-based cell-penetrating peptide increases the efficacy of photodynamic therapy. ChemMedChem 2006, 1, 458–463. [Google Scholar]
- Srinivasan, D.; Muthukrishnan, N.; Johnson, G.A.; Erazo-Oliveras, A.; Lim, J.; Simanek, E.E.; Pellois, J.P. Conjugation to the cell-penetrating peptide TAT potentiates the photodynamic effect of carboxytetramethylrhodamine. PLoS One 2011, 6, e17732. [Google Scholar]
- Wang, J.T.; Giuntini, F.; Eggleston, I.M.; Bown, S.G.; MacRobert, A.J. Photochemical internalisation of a macromolecular protein toxin using a cell penetrating peptide-photosensitiser conjugate. J. Control. Release 2012, 157, 305–313. [Google Scholar] [Green Version]
- Barnett, E.M.; Elangovan, B.; Bullok, K.E.; Piwnica-Worms, D. Selective cell uptake of modified Tat peptide-fluorophore conjugates in rat retina in ex vivo and in vivo models. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2589–2595. [Google Scholar] [CrossRef]
- Redmond, R.W.; Kochevar, I.E. Spatially resolved cellular responses to singlet oxygen. Photochem. Photobiol. 2006, 82, 1178–1186. [Google Scholar] [CrossRef]
- Oliveira, S.; Fretz, M.M.; Hogset, A.; Storm, G.; Schiffelers, R.M. Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA. Biochim. Biophys. Acta 2007, 1768, 1211–1217. [Google Scholar] [CrossRef]
- Endoh, T.; Ohtsuki, T. Cellular siRNA delivery using TatU1A and photo-induced RNA interference. Methods Mol. Biol. 2010, 623, 271–281. [Google Scholar] [CrossRef]
- Muthukrishnan, N.; Johnson, G.A.; Lim, J.; Simanek, E.E.; Pellois, J.P. TAT-mediated photochemical internalization results in cell killing by causing the release of calcium into the cytosol of cells. Biochim. Biophys. Acta 2012, 11, 1734–1743. [Google Scholar]
- Saggu, S.; Hung, H.I.; Quiogue, G.; Lemasters, J.J.; Nieminen, A.L. Lysosomal signaling enhances mitochondria-mediated photodynamic therapy in A431 cancer cells: role of iron. Photochem. Photobiol. 2012, 88, 461–468. [Google Scholar]
- Berg, K.; Dietze, A.; Kaalhus, O.; Hogset, A. Site-specific drug delivery by photochemical internalization enhances the antitumor effect of bleomycin. Clin. Cancer Res. 2005, 11, 8476–8485. [Google Scholar] [CrossRef]
- Nishiyama, N.; Iriyama, A.; Jang, W.D.; Miyata, K.; Itaka, K.; Inoue, Y.; Takahashi, H.; Yanagi, Y.; Tamaki, Y.; Koyama, H.; Kataoka, K. Light-induced gene transfer from packaged DNA enveloped in a dendrimeric photosensitizer. Nat. Mater. 2005, 4, 934–941. [Google Scholar] [CrossRef]
- Selbo, P.K.; Sivam, G.; Fodstad, O.; Sandvig, K.; Berg, K. In vivo documentation of photochemical internalization, a novel approach to site specific cancer therapy. Int. J. Cancer 2001, 92, 761–766. [Google Scholar] [CrossRef]
- Berg, K.; Selbo, P.K.; Weyergang, A.; Dietze, A.; Prasmickaite, L.; Bonsted, A.; Engesaeter, B.O.; Angell-Petersen, E.; Warloe, T.; Frandsen, N.; Hogset, A. Porphyrin-related photosensitizers for cancer imaging and therapeutic applications. J. Microsc. 2005, 218, 133–147. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.-Y.; Pellois, J.-P. Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges. Pharmaceuticals 2012, 5, 1177-1209. https://doi.org/10.3390/ph5111177
Erazo-Oliveras A, Muthukrishnan N, Baker R, Wang T-Y, Pellois J-P. Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges. Pharmaceuticals. 2012; 5(11):1177-1209. https://doi.org/10.3390/ph5111177
Chicago/Turabian StyleErazo-Oliveras, Alfredo, Nandhini Muthukrishnan, Ryan Baker, Ting-Yi Wang, and Jean-Philippe Pellois. 2012. "Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges" Pharmaceuticals 5, no. 11: 1177-1209. https://doi.org/10.3390/ph5111177
APA StyleErazo-Oliveras, A., Muthukrishnan, N., Baker, R., Wang, T. -Y., & Pellois, J. -P. (2012). Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges. Pharmaceuticals, 5(11), 1177-1209. https://doi.org/10.3390/ph5111177