Integration of Ligand-Based Drug Screening with Structure-Based Drug Screening by Combining Maximum Volume Overlapping Score with Ligand Docking

Abstract
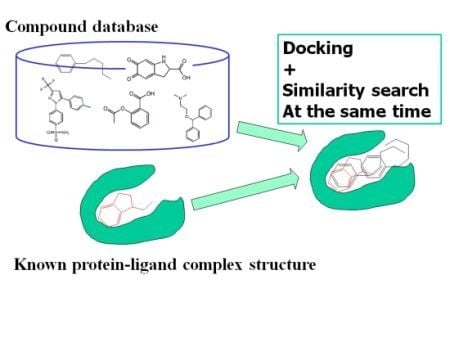
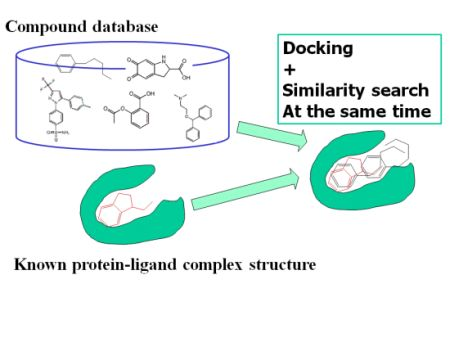
:
1. Introduction

2. Results and Discussion
2.1. Theoretical Background







2.2. Examination of Used Parameters and Evaluation of the Combined MVO with Docking Method

{kind=link}
{kind=link}
| Damping factor | 1 | 1 | 1 | 1 | 1 | 1 | 0.95 | 0.9 | 0.85 | 0.8 | MCS |
| λ | 0.0 | 0.3 | 0.5 | 0.7 | 0.8 | 1.0 | 0.5 | 0.5 | 0.5 | 0.5 | |
| 18gs | 90.2 | 90.3 | 90.4 | 90.4 | 92.7 | 67.7 | 90.5 | 94.7 | 96.2 | 87.3 | 72.7 |
| 1aid | 100.0 | 100.0 | 99.8 | 99.5 | 99.0 | 93.5 | 97.6 | 99.0 | 98.8 | 99.9 | 32.9 |
| 1cbx | 100.0 | 100.0 | 100.0 | 100.0 | 97.0 | 10.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| 1cox | 75.6 | 75.5 | 70.2 | 75.6 | 69.5 | 66.3 | 83.1 | 76.4 | 75.2 | 69.5 | 54.6 |
| 1cps | 97.0 | 99.0 | 99.0 | 95.0 | 88.0 | 73.0 | 98.0 | 97.0 | 97.0 | 99.0 | 100.0 |
| 1gcz | 55.6 | 55.9 | 60.3 | 59.5 | 63.5 | 67.1 | 61.0 | 65.5 | 65.2 | 61.3 | 43.7 |
| 1hpx | 100.0 | 100.0 | 99.9 | 99.9 | 99.8 | 89.2 | 99.2 | 100.0 | 100.0 | 100.0 | 62.3 |
| 1ivp | 100.0 | 99.9 | 99.6 | 99.7 | 99.7 | 96.4 | 99.9 | 100.0 | 100.0 | 99.9 | 67.5 |
| 1pxx | 71.5 | 67.2 | 69.2 | 70.6 | 62.3 | 65.8 | 70.5 | 67.7 | 71.0 | 68.9 | 58.4 |
| 1tlp | 91.2 | 90.9 | 89.8 | 89.4 | 89.9 | 49.7 | 88.6 | 90.0 | 89.1 | 89.5 | 53.0 |
| 1tmn | 84.2 | 84.5 | 81.4 | 80.0 | 79.4 | 59.6 | 84.0 | 89.0 | 88.4 | 90.2 | 52.5 |
| 2gss | 91.6 | 90.2 | 89.0 | 87.1 | 90.3 | 71.5 | 91.4 | 81.3 | 92.3 | 90.5 | 41.8 |
| 2tmn | 90.8 | 92.2 | 92.0 | 90.6 | 90.3 | 36.8 | 91.4 | 92.2 | 90.6 | 91.5 | 55.4 |
| 3cpa | 99.0 | 99.0 | 99.0 | 99.0 | 97.0 | 88.0 | 100.0 | 100.0 | 100.0 | 99.0 | 100.0 |
| 3pgh | 70.7 | 70.5 | 68.4 | 69.1 | 65.6 | 61.0 | 54.8 | 64.3 | 69.9 | 65.0 | 79.5 |
| 3pgt | 90.4 | 88.0 | 89.2 | 92.6 | 91.3 | 81.8 | 91.1 | 92.5 | 86.6 | 88.9 | 83.2 |
| 4cox | 66.7 | 68.7 | 63.2 | 64.9 | 67.0 | 62.6 | 68.6 | 76.4 | 79.5 | 73.5 | 56.7 |
| 6cox | 81.5 | 79.6 | 78.4 | 81.7 | 81.5 | 41.7 | 87.9 | 68.4 | 77.8 | 76.2 | 54.6 |
| Average of AUC | 86.5 | 86.2 | 85.5 | 85.8 | 84.7 | 65.7 | 86.5 | 86.3 | 87.6 | 86.1 | 64.9 |
| of AUC | 13.0 | 13.3 | 13.5 | 12.8 | 13.1 | 21.0 | 13.5 | 12.9 | 11.4 | 13.1 | 19.8 |
| 1% hit ratio | 25.09 | 29.14 | 32.27 | 30.04 | 24.04 | 27.09 | 31.30 | 25.07 | 26.63 | 29.50 | 19.6 |
| Damping factor | 1 | 1 | 1 | 1 | 1 | 1 | 0.95 | 0.9 | 0.85 | 0.8 | MSC |
| λ | 0 | 0.3 | 0.5 | 0.65 | 0.8 | 1 | 0.5 | 0.5 | 0.5 | 0.5 | |
| 18gs | 68.2 | 64.5 | 63.0 | 59.0 | 59.9 | 35.1 | 72.3 | 73.8 | 65.9 | 67.7 | 74.7 |
| 1aid | 85.1 | 86.5 | 78.1 | 77.4 | 75.2 | 69.4 | 74.2 | 71.0 | 77.8 | 77.9 | 45.3 |
| 1cbx | 100.0 | 100.0 | 100.0 | 100.0 | 98.0 | 2.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| 1cox | 29.2 | 37.7 | 43.4 | 62.0 | 64.3 | 52.5 | 48.4 | 40.3 | 47.4 | 12.5 | 67.3 |
| 1cps | 98.0 | 99.0 | 99.0 | 90.0 | 72.0 | 31.0 | 99.0 | 96.0 | 97.0 | 100.0 | 100.0 |
| 1gcz | 27.3 | 30.9 | 36.4 | 43.9 | 56.1 | 49.4 | 36.0 | 38.6 | 40.3 | 31.1 | 63.3 |
| 1hpx | 94.0 | 94.9 | 87.4 | 90.0 | 88.5 | 56.4 | 85.1 | 88.1 | 84.6 | 91.4 | 58.6 |
| 1ivp | 88.7 | 88.7 | 84.5 | 83.4 | 81.6 | 70.7 | 87.7 | 88.4 | 81.8 | 84.1 | 63.0 |
| 1pxx | 21.5 | 25.3 | 32.6 | 43.0 | 44.7 | 47.8 | 22.0 | 16.4 | 36.2 | 27.9 | 76.1 |
| 1tlp | 86.5 | 85.4 | 85.2 | 82.0 | 72.7 | 29.0 | 87.4 | 89.0 | 82.2 | 87.8 | 63.7 |
| 1tmn | 66.6 | 66.2 | 61.0 | 47.4 | 40.1 | 29.4 | 83.0 | 87.1 | 59.2 | 83.2 | 55.3 |
| 2gss | 67.8 | 69.0 | 68.0 | 61.9 | 54.7 | 44.8 | 71.8 | 68.4 | 67.5 | 62.0 | 68.2 |
| 2tmn | 87.4 | 86.5 | 86.7 | 83.3 | 78.6 | 21.8 | 88.8 | 88.7 | 87.6 | 88.9 | 81.7 |
| 3cpa | 100.0 | 100.0 | 100.0 | 100.0 | 99.0 | 83.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| 3pgh | 34.5 | 43.9 | 50.4 | 57.4 | 64.4 | 42.7 | 25.4 | 30.3 | 51.6 | 25.8 | 95.7 |
| 3pgt | 71.6 | 69.6 | 69.3 | 65.6 | 64.5 | 61.7 | 70.7 | 72.2 | 66.3 | 65.3 | 86.1 |
| 4cox | 20.5 | 22.5 | 25.5 | 36.5 | 46.4 | 48.6 | 26.3 | 26.9 | 29.9 | 34.4 | 75.6 |
| 6cox | 37.8 | 41.6 | 51.4 | 66.7 | 74.1 | 28.2 | 50.1 | 30.4 | 53.2 | 20.5 | 67.3 |
| Average of AUC | 65.8 | 67.4 | 67.9 | 69.4 | 68.6 | 44.6 | 68.2 | 67.0 | 68.2 | 64.5 | 74.6 |
| of AUC | 28.5 | 26.5 | 23.3 | 19.2 | 16.5 | 19.3 | 26.0 | 27.6 | 21.4 | 29.9 | 15.9 |
| 1% hit ratio | 13.5 | 13.6 | 17.7 | 18.4 | 18.7 | 9.0 | 18.4 | 16.0 | 20.0 | 20.0 | 28.9 |
| Method | Combined MVO docking | MCS | ||
| λ | 0 | 0.5 | 1 | |
| 4cox | 63.2 | 63.4 | 47.0 | 63.6 |
| 6cox | 51.6 | 53.8 | 42.9 | 73.8 |
| 3ert | 73.3 | 72.0 | 63.4 | 91.2 |
| 3erd | 57.4 | 57.7 | 52.3 | 94.1 |
| 1hpv | 43.1 | 42.3 | 64.0 | 68.9 |
| 1htf | 50.1 | 51.6 | 53.2 | 15.4 |
| 1etr | 63.5 | 59.6 | 45.1 | 86.7 |
| 1ets | 60.1 | 54.7 | 44.1 | 75.5 |
| 1tng | 74.7 | 73.0 | 37.2 | 55.6 |
| 1tnh | 75.5 | 68.5 | 36.4 | 58.0 |
| Average | 61.3 | 59.7 | 48.6 | 68.3 |
| 10.5 | 9.2 | 9.2 | 21.7 | |
| 1% hit ratio | 6.3 | 4.2 | 0.6 | 10.0 |
| PDB ID | Scaffold of ligand | RMSD(Å) (protein) | RMSD (Å)(ligand) | ||
| λ=0 | λ =0.5 | λ =1 | |||
| 1ere | Estrogen (steroid) | 0.00 | 6.90 | 6.60 | 6.22 |
| 1l2i | Tetahydrochrysene | 0.40 | 2.43 | 0.65 | 3.49 |
| 3uuc | Bisphenol | 0.56 | 5.23 | 4.65 | 1.12 |
| 3erd | Triphenylethylene | 0.61 | 2.90 | 2.84 | 4.12 |
| 2iok | Indole | 0.78 | 2.97 | 2.09 | 6.17 |
| 1err | Benzothiophen | 0.79 | 6.21 | 6.15 | 9.85 |
| 1r5k | Triphenylethylene | 0.79 | 7.00 | 6.50 | 7.60 |
| 1yin | Chromane | 1.25 | 3.05 | 3.03 | 7.52 |
| 1sj0 | Benzoxathin | 1.29 | 7.52 | 7.61 | 6.74 |
| 2ouz | Tetrahydronaphthalen | 1.30 | 8.48 | 8.39 | 5.92 |
| 1xp9 | Benzoxathin | 1.31 | 2.24 | 0.98 | 6.57 |
| 1xp6 | Benzoxathin | 1.32 | 2.84 | 3.61 | 9.42 |
| 1xpc | Benzoxathin | 1.33 | 1.72 | 2.54 | 8.87 |
| 1xp1 | Benzoxathin | 1.34 | 2.45 | 2.64 | 9.16 |
| 1yim | Chromane | 1.37 | 2.67 | 2.36 | 5.20 |
| 2iog | Indole | 1.49 | 7.19 | 7.59 | 9.39 |
| 3ert | Triphenylethylene | 1.57 | 2.68 | 2.62 | 8.15 |
| Averaged RMSD (Å) | 4.38 | 4.17 | 6.79 | ||
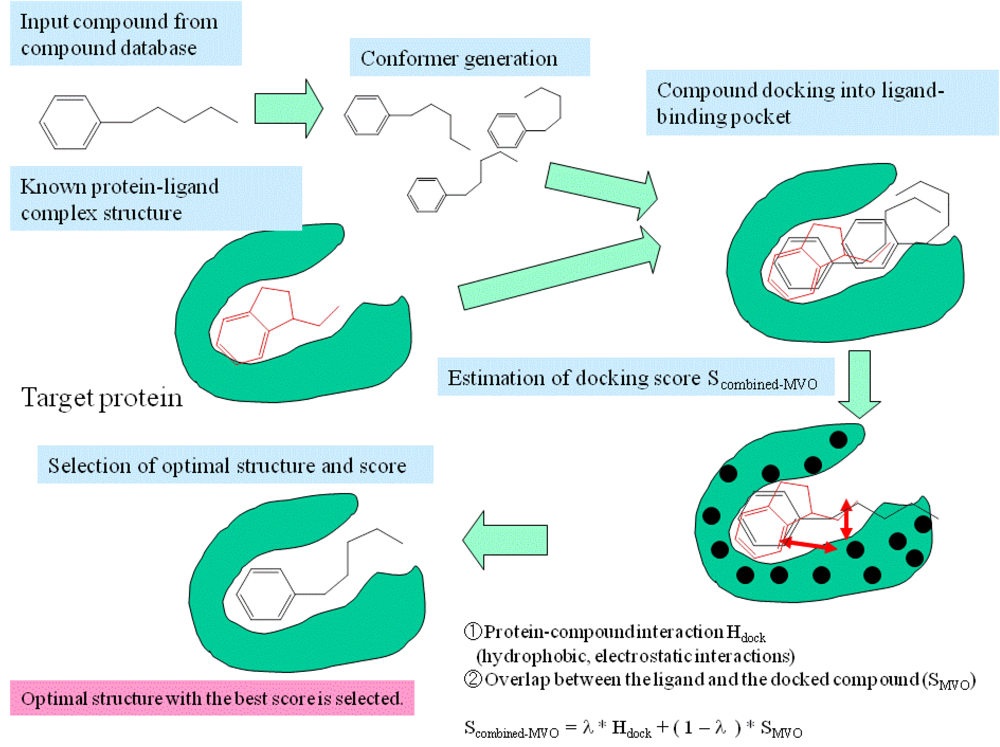
3. Methods: Combining MVO with the Docking Method

- Step 1
- The pocket is indicated by the known ligand coordinates, and the potential energy grids were generated around the ligand-binding pocket.
- Step 2
- Electrostatic potential field on the accessible surface of the receptor is calculated to find a total of 30 potential minima and maxima. Also, hydrophobic potential is calculated by using a methane probe to find those 30 potential minima. Triangles are generated to connect these points; the data regarding these triangles are recorded in a hash table.
- Step 3
- The program reads a compound of the database and then generates its conformers. The dihedral angles are randomly incremented every 120 degrees.
- Step 4
- The global search program chooses any three atoms of the compound and superimposes the compound onto the receptor surface according to the geometric hash method. The Scombined-MVO score is then evaluated.
- Step 5
- Starting from the initial coordinate generated in step 4, the compound coordinates reaches the optimal complex structure using the steepest descent method to minimize the Scombined-MVO score with the grid potential of the receptor force field and the known-ligand coordinates. The AMBER-type molecular force field is used.
4. Preparation of Materials
5. Conclusions
Acknowledgements
References
- van de Waterbeemd, H.; Testa, B.; Folkers, G. Computer-Assisted Lead Finding and Optimization –Current Tools for Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 1997. [Google Scholar]
- Leach, A.R. Molecular Modeling–Principles and Applications, 2nd ed; Pearson Education Limited: Edinburgh Gate, UK, 2001; pp. 668–687. [Google Scholar]
- Richards, W.G.; Robinson, D.D. Rational Drug Design; Truhlar, D.G., Howe, W.J., Hopfinger, A.J., Blaney, J., Dammkoehler, R.A., Eds.; Springer-Verlag: New York, NY, USA, 1999; pp. 39–49. [Google Scholar]
- Pickett, S. Protein-Ligand Interactions from Molecular Recognition to Drug Design–Methods and Principles in Medicinal Chemistry; Boehm, H.J., Schneider, G., Mannhold, R., Kubinyi, H., Folkers, G., Eds.; Wiley-VCH: Weinheim, Germany, 2003; pp. 88–91. [Google Scholar]
- Pearlman, R.S.; Smith, K.M. Metric validation and the receptor-relevant subspace concept. J. Chem. Inf. Compt. Sci. 1999, 39, 28–35. [Google Scholar]
- Fukunishi, Y.; Nakamura, H. Prediction of protein-ligand complex by docking software guided by other complex structures. J. Mol. Graph. Model. 2008, 26, 1030–1033. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Nakamura, H. A new method for in silico drug screening and similarity search using molecular dynamics maximum volume overlap (MD-MVO) method. J. Mol. Graphics Mod. 2009, 27, 628–636. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar]
- Jones, G.; Willet, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins 1990, 8, 195–202. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM: a new method for structure modeling and design: application to docking and structure prediction from the disordered native conformation. J. Compt. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Mikami, Y.; Nakamura, H. Similarities among receptor pockets and among compounds: Analysis and application to in silico ligand screening. J. Mol. Graphics Mod. 2005, 24, 34–45. [Google Scholar] [CrossRef]
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J. Comput. Aided. Mol. Des. 2012, 26, 675–686. [Google Scholar]
- Spitzer, R.; Jain, A.N. Surflex-Dock: docking benchmarks and real-world application. J. Comput. Aided. Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef]
- Schneider, N.; Hindle, S.; Lange, G.; Klein, R.; Albrecht, J. Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J. Comput. Aided Mol. Des. 2012, 26, 701–723. [Google Scholar]
- Novikow, F.N.; Stroylov, V.S.; Zeifman, A.A.; Stroganov, O.V.; Kulkov, V. Lead Finder docking and virtual screening evaluation with Astex and DUD test sets. J. Comput. Aided Mol. Des. 2012, 26, 725–735. [Google Scholar]
- Liebeschuetz, J.W.; Cole, J.C.; orb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Aided Mol. Des. 2012, 26, 737–748. [Google Scholar]
- Brozell, S.R.; Mukherjee, S.; Balius, T.E.; Roe, D.R.; Case, D.A. Evaluation of DOCK 6 as a pose generation and database enrichment tool. J. Comput. Aided Mol. Des. 2012, 26, 749–773. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar]
- Repasky, M.P.; Murphy, R.B.; Banks, J.L.; Greenwood, J.R.; Tubert-brohman, I. Docking performance of the glide program as evaluated on the Astex and DUD datasets: a complete set of glide SP results and selected results for a new scoring function integrating WaterMap and glide. J. Comput. Aided Mol. Des. 2012, 26, 787–799. [Google Scholar] [CrossRef]
- Christofferson, A.J.; Huang, N. Computational Drug Discovery and Design; Humana Press: New York, NY, USA, 2012; pp. 187–195. [Google Scholar]
- Fukunishi, Y.; Kubota, S.; Nakamura, H. Noise reduction method for molecular interaction energy: application to in silico drug screening and in silico target protein screening. J. Chem. Info. Mod. 2006, 46, 2071–2084. [Google Scholar] [CrossRef]
- Fukunishi, Y.; Sugihara, Y.; Mikami, Y.; Sakai, K.; Kusudo, H.; Nakamura, H. Advanced in-silico drug screeing to achieve high hit ratio−development of 3D-compound database. Synthesiology 2009, 2, 64–72. [Google Scholar]
- Cosconati, S.; Marinelli, L.; Leva, F.S.D.; Pietra, V.L.; Simone, A.D.; Mancini, F.; Andrisano, V.; Novellino, E.; Goodsell, D.S.; Olson, A.J. Protein flexibility in virtual screening: the BACE-1 case study. J. Chem. Inf. Model. 2012, 25, 2697–2704. [Google Scholar]
- Rueda, M.; Totrov, M.; Abagyan, R. ALiBERO: evolving a team of complementary pocket conformations rather than a single leader. J. Chem. Inf. Model. 2012, 25, 2705–2714. [Google Scholar]
- Wada, M.; Kanamori, E.; Nakamura, H.; Fukunishi, Y. Selection of in-silico drug screening results for G-protein-coupled receptors by using universal active probe. J. Chem. Inf. Model. 2011, 51, 2398–2407. [Google Scholar] [CrossRef]
- Kawabata, T. Build-up algorithm for atomic correspondence between chemical structures. J. Chem. Info. Mod. 2011, 51, 1775–1787. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar]
- Fukunishi, Y.; Nakamura, H. Improvement of protein-compound docking scores by using amino-acid sequence similarities of proteins. J. Chem. Info. Mod. 2008, 48, 148–156. [Google Scholar]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.III.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Merz, K.M.; Wang, B.; Pearlman, D.A.; Crowley, M.; Brozell, S.; Tsui, V.; Gohlke, H.; Mongan, J.; Hornak, V.; Cui, G.; Beroza, P.; Schafmeister, C.; Caldwell, J.W.; Ross, W.S.; Kollman, P.A. AMBER 8; University of California: San Francisco, CA, 2004. [Google Scholar]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978, 3181–3184. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fukunishi, Y.; Nakamura, H. Integration of Ligand-Based Drug Screening with Structure-Based Drug Screening by Combining Maximum Volume Overlapping Score with Ligand Docking. Pharmaceuticals 2012, 5, 1332-1345. https://doi.org/10.3390/ph5121332
Fukunishi Y, Nakamura H. Integration of Ligand-Based Drug Screening with Structure-Based Drug Screening by Combining Maximum Volume Overlapping Score with Ligand Docking. Pharmaceuticals. 2012; 5(12):1332-1345. https://doi.org/10.3390/ph5121332
Chicago/Turabian StyleFukunishi, Yoshifumi, and Haruki Nakamura. 2012. "Integration of Ligand-Based Drug Screening with Structure-Based Drug Screening by Combining Maximum Volume Overlapping Score with Ligand Docking" Pharmaceuticals 5, no. 12: 1332-1345. https://doi.org/10.3390/ph5121332
APA StyleFukunishi, Y., & Nakamura, H. (2012). Integration of Ligand-Based Drug Screening with Structure-Based Drug Screening by Combining Maximum Volume Overlapping Score with Ligand Docking. Pharmaceuticals, 5(12), 1332-1345. https://doi.org/10.3390/ph5121332