cGMP-Dependent Protein Kinase Inhibitors in Health and Disease


Abstract
:1. Introduction
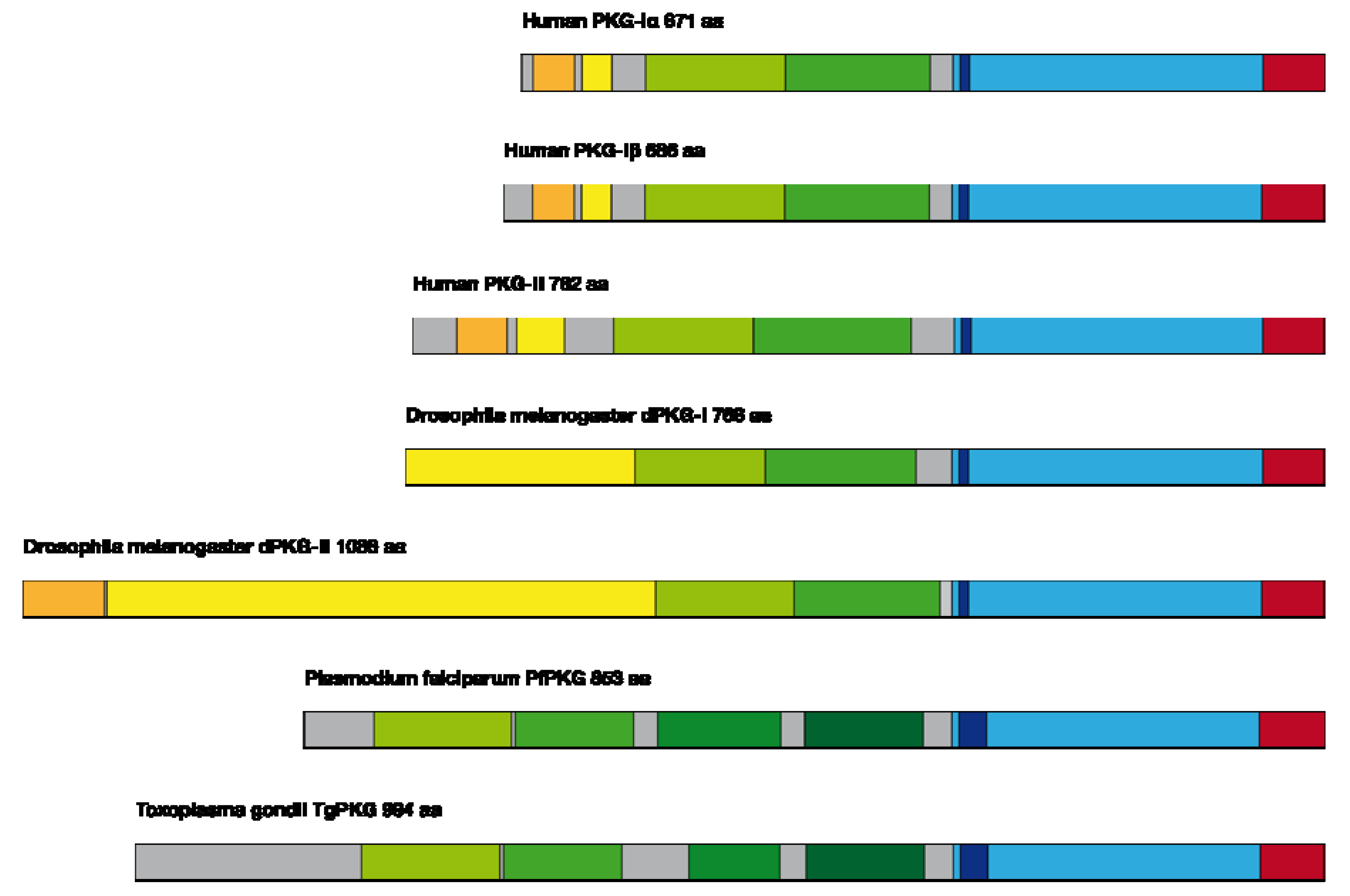
- amino-terminus with a leucine-zipper (important e.g., for homodimerization and targeting) and the autoinhibitory domain.
- regulatory domain with a high- and low-affinity binding site for cGMP (important for the activation of the enzyme).
- catalytic domain for ATP binding, which catalyses the transfer of the phosphate residue to the serine/threonine motif.
2. PKG Inhibitors
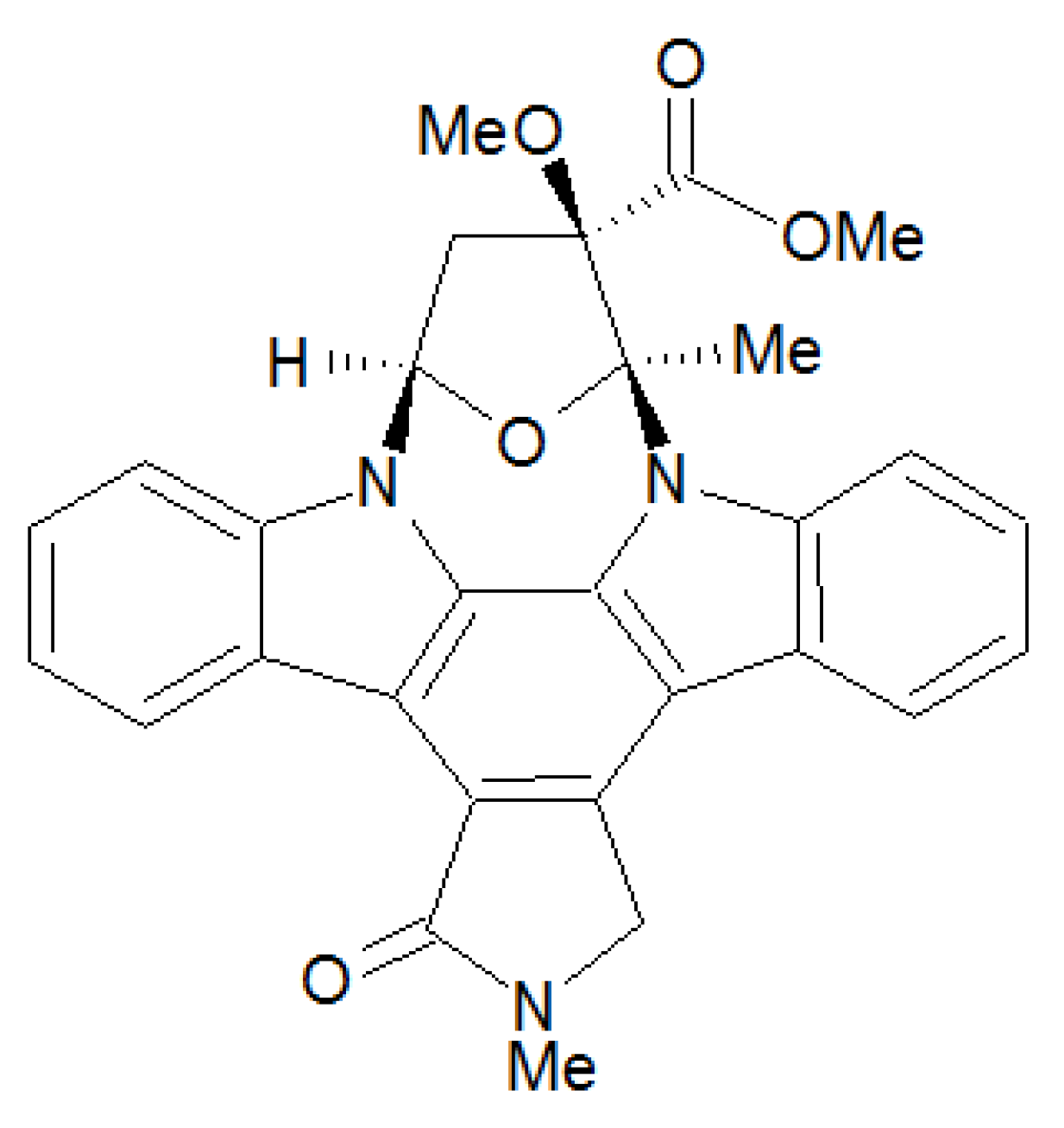
2.1. Cyclic Nucleotide Analogs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors | PKG-Iα | PKG-Iβ | PKG-II | PKA-II | Ref. |
|---|---|---|---|---|---|
| Ki (µM) | Ki (µM) | Ki (µM) | Ki (µM) | ||
| (Rp)-cGMP-S | 20 | 15 | 0.5 | 20 | [22,25] |
| (Rp)-8-Br-cGMP-S | 3.7 | 15 | - | 20 | [22] |
| (Rp)-8-Br-PET-cGMP-S | 0.035 | 0.03 | 0.45-0.9 | 11 | [3,10,22,24,35] |
| (Rp)-8-pCPT-cGMP-S | 0.5 | 0.45-0.6 | 0.29-0.7 | 8.3 | [9,10,22] |
| KT-5823 | 0.23 | - | - | > 10 | [23,28] |
| H-7 | 5.8 | - | - | 3 | [1,28] |
| H-8 | 0.5 | - | - | 1.2 | [1,28] |
| H-9 | 0.9 | - | - | 1.9 | [1,28] |
| H-89 | 0.48-0.5 | - | - | 0.05 | [1,23,28] |
| W45 | 0.49-1.15 | - | - | 559 | [2,30] |
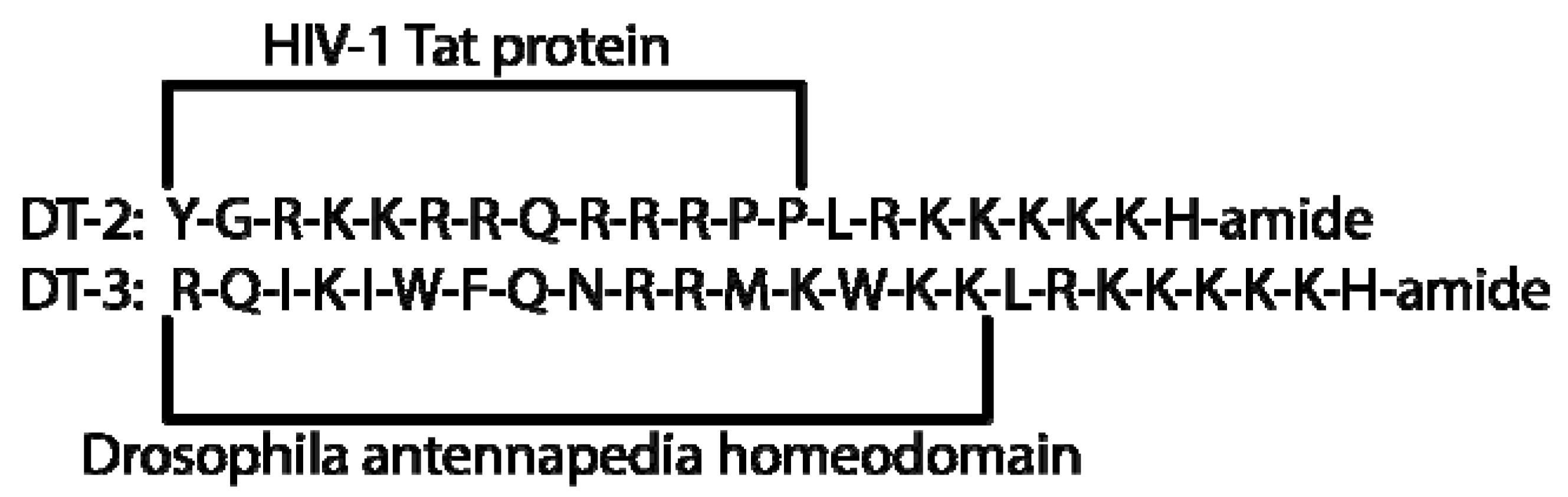
| DT-2 | 0.012 | - | - | 12.7-20.3 | [2,30] |
| DT-3 | 0.025 | - | - | 493 | [30] |
| (D)-DT-2 | 0.0008 | - | - | 8.7-15.3 | [34] |
2.2. K-Series Inhibitors

2.3. H-Series Inhibitors

2.4. W-Series Inhibitors

2.5. DT Inhibitors

2.6. Coccidian PKG Inhibitor

3. PKG-Inhibition as a Potential Therapeutic Target
3.1. PKG in Smooth Muscle Organs
3.1.1. PKG in Vascular Relaxation
3.1.2. PKG in Vascular Remodelling
3.1.3. PKG in the intestine
3.2. PKG in the Bone
3.2.1. PKG in Bone Development
3.3. PKG Signaling in Cancer
3.3.1. PKG-Iα in Lung Cancer
3.3.2. PKG in Colorectal Carcinoma
3.3.3. PKG in Breast Cancer
3.4. PKG Provides a Target for Parasite Treatment
3.4.1. Interspecies Differences in PKG

3.4.2. PKG: A Novel Target in Malaria Therapy

4. Conclusions
Acknowledgments
Conflict of Interest
References
- Ono-Saito, N.; Niki, I.; Hidaka, H. H-series protein kinase inhibitors and potential clinical applications. Pharmacol. Ther. 1999, 82, 123–131. [Google Scholar] [CrossRef]
- Pinkse, M.W.; Rijkers, D.T.; Dostmann, W.R.; Heck, A.J. Mode of action of cGMP-dependent protein kinase-specific inhibitors probed by photoaffinity cross-linking mass spectrometry. The J. Biol. Chem. 2009, 284, 16354–16368. [Google Scholar] [CrossRef]
- Smolenski, A.; Burkhardt, A.M.; Eigenthaler, M.; Butt, E.; Gambaryan, S.; Lohmann, S.M.; Walter, U. Functional analysis of cGMP-dependent protein kinases I and II as mediators of NO/cGMP effects. Naunyn-Schmied. Arch. Pharmacol. 1998, 358, 134–139. [Google Scholar] [CrossRef]
- Hofmann, F. The biology of cyclic GMP-dependent protein kinases. J. Biol. Chem. 2005, 280, 1–4. [Google Scholar]
- Hofmann, F.; Ammendola, A.; Schlossmann, J. Rising behind NO: cGMP-dependent protein kinases. J. Cell Sci. 2000, 113, 1671–1676. [Google Scholar]
- Hofmann, F.; Bernhard, D.; Lukowski, R.; Weinmeister, P. cGMP regulated protein kinases (cGK). Handb. Exp. Pharm. 2009, 137–162. [Google Scholar]
- Lee, J.H.; Li, S.; Liu, T.; Hsu, S.; Kim, C.; Woods, V.L., Jr.; Casteel, D.E. The amino terminus of cGMP-dependent protein kinase Ibeta increases the dynamics of the protein's cGMP-binding pockets. Int. J. Mass. Spectrom. 2011, 302, 44–52. [Google Scholar]
- Busch, J.L.; Bessay, E.P.; Francis, S.H.; Corbin, J.D. A conserved serine juxtaposed to the pseudosubstrate site of type I cGMP-dependent protein kinase contributes strongly to autoinhibition and lower cGMP affinity. J. Biol. Chem. 2002, 277, 34048–34054. [Google Scholar]
- Pohler, D.; Butt, E.; Meissner, J.; Muller, S.; Lohse, M.; Walter, U.; Lohmann, S.M.; Jarchau, T. Expression, purification, and characterization of the cGMP-dependent protein kinases I beta and II using the baculovirus system. FEBS Lett. 1995, 374, 419–425. [Google Scholar] [CrossRef]
- Poppe, H.; Rybalkin, S.D.; Rehmann, H.; Hinds, T.R.; Tang, X.B.; Christensen, A.E.; Schwede, F.; Genieser, H.G.; Bos, J.L.; Doskeland, S.O.; Beavo, J.A.; Butt, E. Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 2008, 5, 277–278. [Google Scholar]
- Richie-Jannetta, R.; Busch, J. L.; Higgins, K.A.; Corbin, J. D.; Francis, S.H. Isolated regulatory domains of cGMP-dependent protein kinase Ialpha and Ibeta retain dimerization and native cGMP-binding properties and undergo isoform-specific conformational changes. J. Biol. Chem. 2006, 281, 6977–6984. [Google Scholar] [CrossRef]
- Wooldridge, A.A.; MacDonald, J.A.; Erdodi, F.; Ma, C.; Borman, M.A.; Hartshorne, D.J.; Haystead, T.A. Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of Serine 695 in response to cyclic nucleotides. J. Biol. Chem. 2004, 279, 34496–34504. [Google Scholar]
- Ellerbroek, S.M.; Wennerberg, K.; Burridge, K. Serine phosphorylation negatively regulates RhoA in vivo. J. Biol. Chem. 2003, 278, 19023–19031. [Google Scholar] [CrossRef]
- Schlossmann, J.; Ammendola, A.; Ashman, K.; Zong, X.; Huber, A.; Neubauer, G.; Wang, G.X.; Allescher, H. D.; Korth, M.; Wilm, M.; Hofmann, F.; Ruth, P. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature 2000, 404, 197–201. [Google Scholar]
- Golin-Bisello, F.; Bradbury, N.; Ameen, N. STa and cGMP stimulate CFTR translocation to the surface of villus enterocytes in rat jejunum and is regulated by protein kinase G. Amer. J. Physiol-Cell Physiol. 2005, 289, C708–C716. [Google Scholar] [CrossRef]
- Chikuda, H.; Kugimiya, F.; Hoshi, K.; Ikeda, T.; Ogasawara, T.; Kamekura, S.; Ogata, N.; Nakamura, K.; Chung, U.I.; Kawaguchi, H. Mutation in cGMP-dependent protein kinase II causes dwarfism in a rat mutant KMI through uncoupling of proliferation and differentiation of chondrocytes. J. Bone Miner. Metab. 2005, 23, 200–204. [Google Scholar] [CrossRef]
- Forte, L.R.; London, R.M.; Krause, W.J.; Freeman, R.H. Mechanisms of guanylin action via cyclic GMP in the kidney. Annu. Rev. Physiol. 2000, 62, 673–695. [Google Scholar] [CrossRef]
- Rangaswami, H.; Marathe, N.; Zhuang, S.; Chen, Y.; Yeh, J.C.; Frangos, J.A.; Boss, G.R.; Pilz, R.B. Type II cGMP-dependent protein kinase mediates osteoblast mechanotransduction. J. Biol. Chem. 2009, 284, 14796–14808. [Google Scholar]
- Gambaryan, S.; Wagner, C.; Smolenski, A.; Walter, U.; Poller, W.; Haase, W.; Kurtz, A.; Lohmann, S.M. Endogenous or overexpressed cGMP-dependent protein kinases inhibit cAMP-dependent renin release from rat isolated perfused kidney, microdissected glomeruli, and isolated juxtaglomerular cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9003–9008. [Google Scholar]
- Schlossmann, J.; Feil, R.; Hofmann, F. Signaling through NO and cGMP-dependent protein kinases. Ann. Med. 2003, 35, 21–27. [Google Scholar]
- Butt, E. cGMP-dependent protein kinase modulators. Handb. Exp. Pharm. 2009, 409–421. [Google Scholar] [CrossRef]
- Schwede, F.; Maronde, E.; Genieser, H.; Jastorff, B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol. Ther. 2000, 87, 199–226. [Google Scholar] [CrossRef]
- Burkhardt, M.; Glazova, M.; Gambaryan, S.; Vollkommer, T.; Butt, E.; Bader, B.; Heermeier, K.; Lincoln, T.M.; Walter, U.; Palmetshofer, A. KT5823 inhibits cGMP-dependent protein kinase activity in vitro but not in intact human platelets and rat mesangial cells. J. Biol. Chem. 2000, 275, 33536–33541. [Google Scholar]
- Butt, E.; Pohler, D.; Genieser, H.G.; Huggins, J.P.; Bucher, B. Inhibition of cyclic GMP-dependent protein kinase-mediated effects by (Rp)-8-bromo-PET-cyclic GMPS. Brit. J. Pharmacol. 1995, 116, 3110–3116. [Google Scholar] [CrossRef]
- Butt, E.; van Bemmelen, M.; Fischer, L.; Walter, U.; Jastorff, B. Inhibition of cGMP-dependent protein kinase by (Rp)-guanosine 3',5'-monophosphorothioates. FEBS Lett. 1990, 263, 47–50. [Google Scholar] [CrossRef]
- Butt, E.; Abel, K.; Krieger, M.; Palm, D.; Hoppe, V.; Hoppe, J.; Walter, U. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J. Biol. Chem. 1994, 269, 14509–14517. [Google Scholar]
- Vaandrager, A.B.; Bot, A.G.; De Jonge, H.R. Guanosine 3',5'-cyclic monophosphate-dependent protein kinase II mediates heat-stable enterotoxin-provoked chloride secretion in rat intestine. Gastroenterology 1997, 112, 437–443. [Google Scholar] [CrossRef]
- Hidaka, H.; Kobayashi, R. Pharmacology of protein kinase inhibitors. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 377–397. [Google Scholar] [CrossRef]
- Engh, R.A.; Girod, A.; Kinzel, V.; Huber, R.; Bossemeyer, D. Crystal structures of catalytic subunit of cAMP-dependent protein kinase in complex with isoquinolinesulfonyl protein kinase inhibitors H7, H8, and H89. Structural implications for selectivity. J. Biol. Chem. 1996, 271, 26157–26164. [Google Scholar]
- Dostmann, W.R.; Taylor, M.S.; Nickl, C.K.; Brayden, J.E.; Frank, R.; Tegge, W.J. Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Ialpha inhibit NO-induced cerebral dilation. Proc. Nat. Acad. Sci. USA 2000, 97, 14772–14777. [Google Scholar] [CrossRef]
- Dostmann, W.R.; Tegge, W.; Frank, R.; Nickl, C.K.; Taylor, M.S.; Brayden, J.E. Exploring the mechanisms of vascular smooth muscle tone with highly specific, membrane-permeable inhibitors of cyclic GMP-dependent protein kinase Ialpha. Pharmacol. Ther. 2002, 93, 203–215. [Google Scholar] [CrossRef]
- Gambaryan, S.; Butt, E.; Kobsar, A.; Geiger, J.; Rukoyatkina, N.; Parnova, R.; Nikolaev, V.O.; Walter, U. The oligopeptide DT-2 is a specific PKG I inhibitor only in vitro, not in living cells. Brit. J. Pharmacol. 2012, 167, 826–838. [Google Scholar] [CrossRef]
- Foley, K.F.; De Frutos, S.; Laskovski, K.E.; Tegge, W.; Dostmann, W. R. Culture conditions influence uptake and intracellular localization of the membrane permeable cGMP-dependent protein kinase inhibitor DT-2. Front Biosci. 2005, 10, 1302–1312. [Google Scholar] [CrossRef]
- Nickl, C.K.; Raidas, S.K.; Zhao, H.; Sausbier, M.; Ruth, P.; Tegge, W.; Brayden, J.E.; Dostmann, W.R. (D)-Amino acid analogues of DT-2 as highly selective and superior inhibitors of cGMP-dependent protein kinase Ialpha. Biochim. Biophys. Acta 2010, 1804, 524–532. [Google Scholar] [CrossRef]
- Vaandrager, A.B.; Edixhoven, M.; Bot, A.G.; Kroos, M.A.; Jarchau, T.; Lohmann, S.; Genieser, H.G.; de Jonge, H.R. Endogenous type II cGMP-dependent protein kinase exists as a dimer in membranes and can Be functionally distinguished from the type I isoforms. J. Biol. Chem. 1997, 272, 11816–11823. [Google Scholar]
- Gurnett, A.M.; Liberator, P.A.; et al. Purification and molecular characterization of cGMP-dependent protein kinase from Apicomplexan parasites. A novel chemotherapeutic target. J. Biol. Chem. 2002, 277, 15913–15922. [Google Scholar] [CrossRef]
- Koeppen, M.; Feil, R.; Siegl, D.; Feil, S.; Hofmann, F.; Pohl, U.; de Wit, C. cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension 2004, 44, 952–955. [Google Scholar] [CrossRef]
- Pfeifer, A.; Klatt, P.; Massberg, S.; Ny, L.; Sausbier, M.; Hirneiss, C.; Wang, G. X.; Korth, M.; Aszodi, A.; Andersson, K.E.; Krombach, F.; Mayerhofer, A.; Ruth, P.; Fassler, R.; Hofmann, F. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998, 17, 3045–3051. [Google Scholar]
- Sausbier, M.; Schubert, R.; Voigt, V.; Hirneiss, C.; Pfeifer, A.; Korth, M.; Kleppisch, T.; Ruth, P.; Hofmann, F. Mechanisms of NO/cGMP-dependent vasorelaxation. Circ. Res. 2000, 87, 825–830. [Google Scholar] [CrossRef]
- Weber, S.; Bernhard, D.; Lukowski, R.; Weinmeister, P.; Worner, R.; Wegener, J. W.; Valtcheva, N.; Feil, S.; Schlossmann, J.; Hofmann, F.; Feil, R. Rescue of cGMP kinase I knockout mice by smooth muscle specific expression of either isozyme. Circ. Res. 2007, 101, 1096–1103. [Google Scholar] [CrossRef]
- Surks, H.K.; Mochizuki, N.; Kasai, Y.; Georgescu, S.P.; Tang, K.M.; Ito, M.; Lincoln, T.M.; Mendelsohn, M.E. Regulation of myosin phosphatase by a specific interaction with cGMP- dependent protein kinase Ialpha. Science 1999, 286, 1583–1587. [Google Scholar] [CrossRef]
- Robertson, B.E.; Schubert, R.; Hescheler, J.; Nelson, M.T. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Amer. J. Physiol. 1993, 265, C299–C303. [Google Scholar]
- Desch, M.; Sigl, K.; Hieke, B.; Salb, K.; Kees, F.; Bernhard, D.; Jochim, A.; Spiessberger, B.; Hocherl, K.; Feil, R.; Feil, S.; Lukowski, R.; Wegener, J.W.; Hofmann, F.; Schlossmann, J. IRAG determines nitric oxide- and atrial natriuretic peptide-mediated smooth muscle relaxation. Cardiovasc. Res. 2010, 86, 496–505. [Google Scholar]
- Taylor, M.S.; Okwuchukwuasanya, C.; Nickl, C.K.; Tegge, W.; Brayden, J.E.; Dostmann, W.R. Inhibition of cGMP-dependent protein kinase by the cell-permeable peptide DT-2 reveals a novel mechanism of vasoregulation. Mol. Pharmacol. 2004, 65, 1111–1119. [Google Scholar] [CrossRef]
- Dey, N.B.; Busch, J.L.; Francis, S.H.; Corbin, J.D.; Lincoln, T.M. Cyclic GMP specifically suppresses Type-Ialpha cGMP-dependent protein kinase expression by ubiquitination. Cell. Signal. 2009, 21, 859–866. [Google Scholar] [CrossRef]
- Ozaki, M.; Kawashima, S.; Yamashita, T.; Hirase, T.; Namiki, M.; Inoue, N.; Hirata, K.; Yasui, H.; Sakurai, H.; Yoshida, Y.; Masada, M.; Yokoyama, M. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J. Clin. Invest. 2002, 110, 331–340. [Google Scholar]
- Shi, W.; Wang, X.; Shih, D.M.; Laubach, V.E.; Navab, M.; Lusis, A.J. Paradoxical reduction of fatty streak formation in mice lacking endothelial nitric oxide synthase. Circulation 2002, 105, 2078–2082. [Google Scholar] [CrossRef]
- Chen, J.; Kuhlencordt, P.J.; Astern, J.; Gyurko, R.; Huang, P.L. Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein e/endothelial nitric oxide synthase double knockout mice. Circulation 2001, 104, 2391–2394. [Google Scholar] [CrossRef]
- Knowles, J.W.; Reddick, R.L.; Jennette, J.C.; Shesely, E.G.; Smithies, O.; Maeda, N. Enhanced atherosclerosis and kidney dysfunction in eNOS(-/-)Apoe(-/-) mice are ameliorated by enalapril treatment. J. Clin. Invest. 2000, 105, 451–458. [Google Scholar] [CrossRef]
- Wong, J.C.; Fiscus, R.R. Protein kinase G activity prevents pathological-level nitric oxide-induced apoptosis and promotes DNA synthesis/cell proliferation in vascular smooth muscle cells. Cardiovasc. Pathol. 2010, 19, e221–e231. [Google Scholar] [CrossRef]
- Wolfsgruber, W.; Feil, S.; Brummer, S.; Kuppinger, O.; Hofmann, F.; Feil, R. A proatherogenic role for cGMP-dependent protein kinase in vascular smooth muscle cells. Proc. Natl. Acad. Sci. US A 2003, 100, 13519–13524. [Google Scholar] [CrossRef]
- Weinmeister, P.; Lukowski, R.; Linder, S.; Traidl-Hoffmann, C.; Hengst, L.; Hofmann, F.; Feil, R. Cyclic guanosine monophosphate-dependent protein kinase I promotes adhesion of primary vascular smooth muscle cells. Mol. Biol. Cell. 2008, 19, 4434–4441. [Google Scholar] [CrossRef]
- Lincoln, T.M.; Wu, X.; Sellak, H.; Dey, N.; Choi, C.S. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front. Biosci. 2006, 11, 356–367. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar]
- Lukowski, R.; Weinmeister, P.; Bernhard, D.; Feil, S.; Gotthardt, M.; Herz, J.; Massberg, S.; Zernecke, A.; Weber, C.; Hofmann, F.; Feil, R. Role of smooth muscle cGMP/cGKI signaling in murine vascular restenosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1244–1250. [Google Scholar] [CrossRef]
- Joshi, C.N.; Martin, D.N.; Fox, J.C.; Mendelev, N.N.; Brown, T.A.; Tulis, D.A. The soluble guanylate cyclase stimulator BAY 41-2272 inhibits vascular smooth muscle growth through the cAMP-dependent protein kinase and cGMP-dependent protein kinase pathways. J. Pharmacol. Exp. Ther. 2011, 339, 394–402. [Google Scholar] [CrossRef]
- Koika, V.; Zhou, Z.; Vasileiadis, I.; Roussos, C.; Finetti, F.; Monti, M.; Morbidelli, L.; Papapetropoulos, A. PKG-I inhibition attenuates vascular endothelial growth factor-stimulated angiogenesis. Vasc. Pharmacol. 2010, 53, 215–222. [Google Scholar] [CrossRef]
- Kim, B.J.; Lee, J.H.; Jun, J.Y.; Chang, I.Y.; So, I.; Kim, K.W. Vasoactive intestinal polypeptide inhibits pacemaker activity via the nitric oxide-cGMP-protein kinase G pathway in the interstitial cells of Cajal of the murine small intestine. Mol. Cells 2006, 21, 337–342. [Google Scholar] [CrossRef]
- Vaandrager, A.B.; Bot, A.G.; Ruth, P.; Pfeifer, A.; Hofmann, F.; De Jonge, H.R. Differential role of cyclic GMP-dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology 2000, 118, 108–114. [Google Scholar]
- Vaandrager, A.B.; Smolenski, A.; Tilly, B.C.; Houtsmuller, A.B.; Ehlert, E.M.; Bot, A.G.; Edixhoven, M.; Boomaars, W.E.; Lohmann, S.M.; de Jonge, H.R. Membrane targeting of cGMP-dependent protein kinase is required for cystic fibrosis transmembrane conductance regulator Cl- channel activation. Proc. Nat. Acad. Sci. USA 1998, 95, 1466–1471. [Google Scholar]
- Pfeifer, A.; Aszodi, A.; Seidler, U.; Ruth, P.; Hofmann, F.; Fassler, R. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science 1996, 274, 2082–2086. [Google Scholar]
- Chikuda, H.; Kugimiya, F.; Hoshi, K.; Ikeda, T.; Ogasawara, T.; Shimoaka, T.; Kawano, H.; Kamekura, S.; Tsuchida, A.; Yokoi, N.; Nakamura, K.; Komeda, K.; Chung, U. I.; Kawaguchi, H. Cyclic GMP-dependent protein kinase II is a molecular switch from proliferation to hypertrophic differentiation of chondrocytes. Gene Dev. 2004, 18, 2418–2429. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Kugimiya, F.; Chikuda, H.; Kamekura, S.; Ikeda, T.; Kawamura, N.; Saito, T.; Shinoda, Y.; Higashikawa, A.; Yano, F.; Ogasawara, T.; Ogata, N.; Hoshi, K.; Hofmann, F.; Woodgett, J.R.; Nakamura, K.; Chung, U.I.; Kawaguchi, H. Phosphorylation of GSK-3beta by cGMP-dependent protein kinase II promotes hypertrophic differentiation of murine chondrocytes. J. Clin. Invest. 2008, 118, 2506–2515. [Google Scholar]
- Rangaswami, H.; Marathe, N.; Zhuang, S.; Chen, Y.; Yeh, J. C.; Frangos, J.A.; Boss, G.R.; Pilz, R.B. Type II cGMP-dependent protein kinase mediates osteoblast mechanotransduction. J. Biol. Chem. 2009, 284, 14796–14808. [Google Scholar]
- Rangaswami, H.; Schwappacher, R.; Tran, T.; Chan, G.C.; Zhuang, S.; Boss, G.R.; Pilz, R.B. Protein kinase G and focal adhesion kinase converge on Src/Akt/beta-catenin signaling module in osteoblast mechanotransduction. J. Biol. Chem. 2012, 287, 21509–21519. [Google Scholar]
- Wong, J.C.; Fiscus, R.R. Essential roles of the nitric oxide (no)/cGMP/protein kinase G type-Ialpha (PKG-Ialpha) signaling pathway and the atrial natriuretic peptide (ANP)/cGMP/PKG-Ialpha autocrine loop in promoting proliferation and cell survival of OP9 bone marrow stromal cells. J. Cell. Biochem. 2011, 112, 829–839. [Google Scholar] [CrossRef]
- Johlfs, M.G.; Fiscus, R.R. Protein kinase G type-Ialpha phosphorylates the apoptosis-regulating protein Bad at serine 155 and protects against apoptosis in N1E-115 cells. Neurochem. Int. 2010, 56, 546–553. [Google Scholar] [CrossRef]
- Leung, E.L.; Wong, J.C.; Johlfs, M.G.; Tsang, B.K.; Fiscus, R.R. Protein kinase G type Ialpha activity in human ovarian cancer cells significantly contributes to enhanced Src activation and DNA synthesis/cell proliferation. Mol. Cancer Res. 2010, 8, 578–591. [Google Scholar] [CrossRef]
- Wong, J.C.; Bathina, M.; Fiscus, R.R. Cyclic GMP/protein kinase G type-Ialpha (PKG-Ialpha) signaling pathway promotes CREB phosphorylation and maintains higher c-IAP1, livin, survivin, and Mcl-1 expression and the inhibition of PKG-Ialpha kinase activity synergizes with cisplatin in non-small cell lung cancer cells. J. Cell. Biochem. 2012, 113, 3587–3598. [Google Scholar] [CrossRef]
- Babykutty, S.; Suboj, P.; Srinivas, P.; Nair, A.S.; Chandramohan, K.; Gopala, S. Insidious role of nitric oxide in migration/invasion of colon cancer cells by upregulating MMP-2/9 via activation of cGMP-PKG-ERK signaling pathways. Clin. Exp. Metastas. 2012, 29, 471–492. [Google Scholar] [CrossRef]
- Hou, Y.; Wong, E.; Martin, J.; Schoenlein, P.V.; Dostmann, W.R.; Browning, D.D. A role for cyclic-GMP dependent protein kinase in anoikis. Cell. signal. 2006, 18, 882–888. [Google Scholar]
- Fallahian, F.; Karami-Tehrani, F.; Salami, S.; Aghaei, M. Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS. J. 2011, 278, 3360–3369. [Google Scholar] [CrossRef]
- Taylor, H.M.; McRobert, L.; Grainger, M.; Sicard, A.; Dluzewski, A.R.; Hopp, C.S.; Holder, A.A.; Baker, D.A. The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryot. Cell 2010, 9, 37–45. [Google Scholar] [CrossRef]
- Hopp, C.S.; Flueck, C.; Solyakov, L.; Tobin, A.; Baker, D.A. Spatiotemporal and Functional Characterisation of the Plasmodium falciparum cGMP-Dependent Protein Kinase. PloS One 2012, 7, e48206. [Google Scholar]
- Wiersma, H.I.; Galuska, S.E.; Tomley, F.M.; Sibley, L.D.; Liberator, P.A.; Donald, R.G. A role for coccidian cGMP-dependent protein kinase in motility and invasion. Int. J. Parasitol. 2004, 34, 369–380. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wolfertstetter, S.; Huettner, J.P.; Schlossmann, J. cGMP-Dependent Protein Kinase Inhibitors in Health and Disease. Pharmaceuticals 2013, 6, 269-286. https://doi.org/10.3390/ph6020269
Wolfertstetter S, Huettner JP, Schlossmann J. cGMP-Dependent Protein Kinase Inhibitors in Health and Disease. Pharmaceuticals. 2013; 6(2):269-286. https://doi.org/10.3390/ph6020269
Chicago/Turabian StyleWolfertstetter, Stefanie, Johannes P. Huettner, and Jens Schlossmann. 2013. "cGMP-Dependent Protein Kinase Inhibitors in Health and Disease" Pharmaceuticals 6, no. 2: 269-286. https://doi.org/10.3390/ph6020269
APA StyleWolfertstetter, S., Huettner, J. P., & Schlossmann, J. (2013). cGMP-Dependent Protein Kinase Inhibitors in Health and Disease. Pharmaceuticals, 6(2), 269-286. https://doi.org/10.3390/ph6020269