1. Introduction
Since the discovery of RNA interference (RNAi), substantial efforts have been directed towards harnessing the sequence-specific silencing potential of small interfering (si)RNAs for therapeutic applications. Despite important advances in chemical modification strategies to enhance the stability of siRNAs in serum and reduce their off-target effects, delivery remains the most significant hurdle to the therapeutic application of siRNAs [
1,
2,
3]. Effective use of RNAi
in vivo depends on the delivery of exogenous siRNAs into the cytoplasm of target cells in quantities sufficient to silence gene expression and elicit the intended therapeutic effect. To this end, extensive research has focused on the development of targeted delivery systems capable of directing the siRNAs to the target cells and facilitating endosomal escape. Several of the existing delivery approaches make use of cell-specific targeting ligands or functional groups covalently conjugated to siRNAs to direct their delivery to a specific cell-type or tissue [
2].
One such approach, based on synthetic RNA ligands (aptamers), has been employed by us and others, to facilitate delivery of siRNAs to the cytoplasm of target cells both
in vitro and
in vivo [
4,
5,
6,
7,
8,
9,
10,
11,
12]. For this approach, aptamers that bind cell-specific receptors are conjugated with partially (one strand only) or fully (both strands) chemically-modified siRNAs in chimeric molecules. The aptamer directs the chimeric RNA (aptamer-siRNA conjugate) to the cells that express the aptamer-targeted receptor on their surface. The chimeric RNA is then internalized, released into the cytoplasm (by a mechanism that remains to be fully understood) and the siRNA is processed by the RNAi machinery, resulting in mRNA knockdown of the siRNA target gene selectively in the targeted cell population. We have pioneered this strategy for systemic administration of therapeutic anti-cancer siRNAs to mice bearing human-derived prostate tumors [
4,
5]. Since its conception, this strategy has been validated
in vivo with systemic administration in xenograft mouse models of prostate cancer [
5,
10,
11] and HIV-infected cells [
9,
12,
13]. While the potential of this approach as a platform technology with broad applicability is substantial, its widespread adoption is contingent on the availability of aptamers to cell-surface receptors capable of entering and delivering their siRNA cargo to the cytoplasm of target cells.
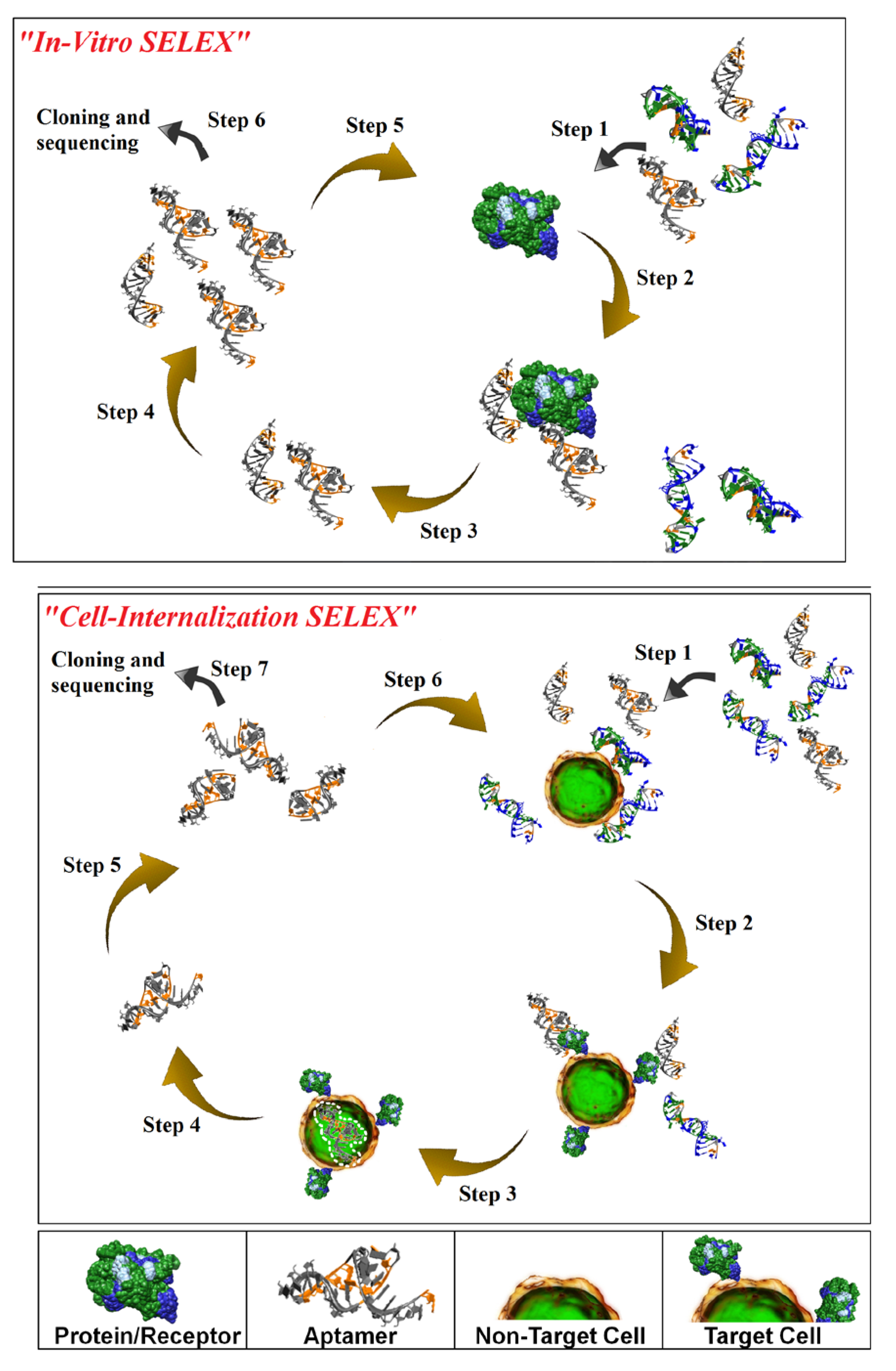
Isolation of aptamers with affinity and specificity for a target of interest involves iterative rounds of affinity purification and amplification via a process termed
in vitro-Systematic Evolution of Ligands by EXponential enrichment (SELEX) [
14,
15] (
Figure 1; top panel). While
in vitro-SELEX results in aptamers with affinities and specificities for their cognate targets comparable to those seen with antibodies, this process does not guarantee the identification of aptamers capable of recognizing their target in the context of the cell membrane and of internalizing into the target cell. We have recently described a novel cell-based selection strategy that we refer to as cell-internalization SELEX for isolating aptamers that internalize upon binding to their cognate receptor [
16,
17,
18] (
Figure 1; bottom panel). Cell-internalization SELEX has several advantages over other selection approaches for targeted therapeutic applications: (1) it favors the isolation of RNAs that bind to receptors in their native state; and (2) it enriches for RNAs capable of entering the target cell. To date, this approach has yielded aptamers capable of internalizing into HER2-positive mammary carcinoma cells [
16], vascular smooth muscle cells [
17] and TrkB-expressing cells [
18]. Importantly, we demonstrated that when conjugated to therapeutic siRNAs, the cell-internalizing aptamers were capable of delivering their cargo to the cytoplasm of the target cells resulting in a robust RNAi response [
16].
Figure 1.
Schematic of in vitro-Systematic Evolution of Ligands by EXponential enrichment (SELEX; upper panel) and Cell-Internalization SELEX methodologies (lower panel). For in vitro-SELEX, a library of RNA molecules is incubated with the target (e.g. recombinant protein) (Step 1) and binders are separated from non-binders (Step 2). Recovered binders (Step 3) are subsequently amplified (Step 4) to generate a pool of molecules that displays high affinity and specificity for the target. For cell-internalization SELEX, a library of RNA molecules is incubated with non-target cells (counter selection) (Step 1) and those RNAs that do not bind (supernatant) are then transferred to the target cells (Step 2). The cell incubations are performed at 37 °C to facilitate cell internalization (Step 3). A stringent wash step (high salt wash, trypsin wash) is then performed to remove unbound and/or surface bound RNA. The internalized RNA is recovered using TRIzol extraction (Step 4) and amplified by PCR (Step 5). The process is repeated several more times to enrich for RNA sequences with cell-specific, internalizing properties (Step 6). The enriched pool of RNA is then subjected to cloning and sequencing (Step 7).
Figure 1.
Schematic of in vitro-Systematic Evolution of Ligands by EXponential enrichment (SELEX; upper panel) and Cell-Internalization SELEX methodologies (lower panel). For in vitro-SELEX, a library of RNA molecules is incubated with the target (e.g. recombinant protein) (Step 1) and binders are separated from non-binders (Step 2). Recovered binders (Step 3) are subsequently amplified (Step 4) to generate a pool of molecules that displays high affinity and specificity for the target. For cell-internalization SELEX, a library of RNA molecules is incubated with non-target cells (counter selection) (Step 1) and those RNAs that do not bind (supernatant) are then transferred to the target cells (Step 2). The cell incubations are performed at 37 °C to facilitate cell internalization (Step 3). A stringent wash step (high salt wash, trypsin wash) is then performed to remove unbound and/or surface bound RNA. The internalized RNA is recovered using TRIzol extraction (Step 4) and amplified by PCR (Step 5). The process is repeated several more times to enrich for RNA sequences with cell-specific, internalizing properties (Step 6). The enriched pool of RNA is then subjected to cloning and sequencing (Step 7).
![Pharmaceuticals 06 00295 g001]()
Although, the cell-internalization SELEX protocol has been useful at identifying several aptamers for siRNA-delivery [
16,
17], this approach does not necessarily select for aptamers that effectively escape endosomes and are released into the cytoplasm of target cells. Modifications to this approach (e.g., inclusion of fractionation steps to separate endosome-bound from cytoplasmic aptamer sequences) that allow for the selective amplification of cytoplasmic-specific aptamers may enable the effective isolation of cytoplasmic-targeting RNA sequences. The current cell-targeted aptamers for siRNA delivery must escape from early or late endosomes prior to entering the lysosome where the presence of nuclease and low pH conditions are likely to result in degradation of the aptamer-siRNA complexes [
19]. Currently, neither the uptake kinetics of the cell-targeted aptamers nor the efficiency of release from endosomes is known.
Arguably, the future of this technology rests on the development of robust assays for the characterization, validation and optimization of cell-targeting aptamers as delivery agents. Here we describe several qualitative assays for assessing cell-specific internalization (microscopy-, microplate- and flow cytometry-based assays) and sub-cellular localization (RNA-RIP assay) of the RNAs. We also describe a novel quantitative assay (“QUSIM”) for measuring the efficiency of aptamer internalization. These methodologies promise to facilitate the development and characterization of cell-internalizing aptamers for delivery of therapeutics (e.g., siRNAs) into their target cells.
3. Experimental
3.1. Cell Culture and Transfection
All cell lines were maintained in NuAire water-jacketed CO
2 incubators at 37 °C with 5% CO
2. N202.1A and N202.1E cells [
34,
35] were generously provided by G. Forni (University of Torino, Italy). N202.1A is a mammary carcinoma clonal cell line derived from a mammary tumor from the HER2/neu transgenic mouse of FVB background. N202.1A cells express high levels of surface HER2/neu, while the N202.1 E clonal cell line has no detectable surface expression of the HER2/neu oncoprotein. N202.1E-h HER2 cells were generated by stably transfecting N202.1E cell line with ErbB2-pHIV7-neo lentiviral construct using Polybrene, kindly provided by Al Klingelhutz (University of Iowa, USA). N202.1A (HER2
+) and N202.1E (HER2
−) cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 20% fetal bovine serum (FBS, HyClone, Logan, UT, USA). N202.1E-h HER2 cells were maintained in medium supplemented with 200 μg/mL G-418. PSMA-non expressing prostate cancer cell line, PC3(PSMA-), and PSMA-expressing prostate cancer cell line, PC3(PSMA+), were maintained according to the supplier’s recommendations (ATCC #CRL-1435) in DMEM/F12 media with 10% FBS. PC-3(PSMA+) were derived from PSMA expressing PC-3 cells (generously provided by Eli Gilboa, University of Miami, USA) by clonally expanding a clone with high PSMA expression and further sorting using PSMA-PE labeled anti-PSMA antibody from MBL. HEK293 (ATCC cat# CRL-1573) and HEK293 cells expressing TrkB were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. HEK293 cells were stably transfected with TrkB plasmid DNA by the lipofectamine method (Invitrogen, Grand Island, NY, USA). To achieve high-expressing TrkB cells, the cells were subjected to an anti-TrkB antibody recognizing the extra cellular domain (Chemicon cat# AB9872), followed by a secondary antibody conjugated to Alexa Fluor 488 (goat anti-rabbit AF 488, Invitrogen cat # A11008), as previously described [
18]. SK-BR-3 breast adenocarcinoma cells (ATCC cat# HTB-30) were cultured in ATCC-formulated McCoy’s 5a Medium Modified (Cat# 30-2021) supplemented with 10% FBS. All cell lines were screened for contamination by mycoplasma, which is known to degrade 2′-fluoro pyrimidine modified RNA aptamers [
36].
3.2. Receptor Cell Surface Expression
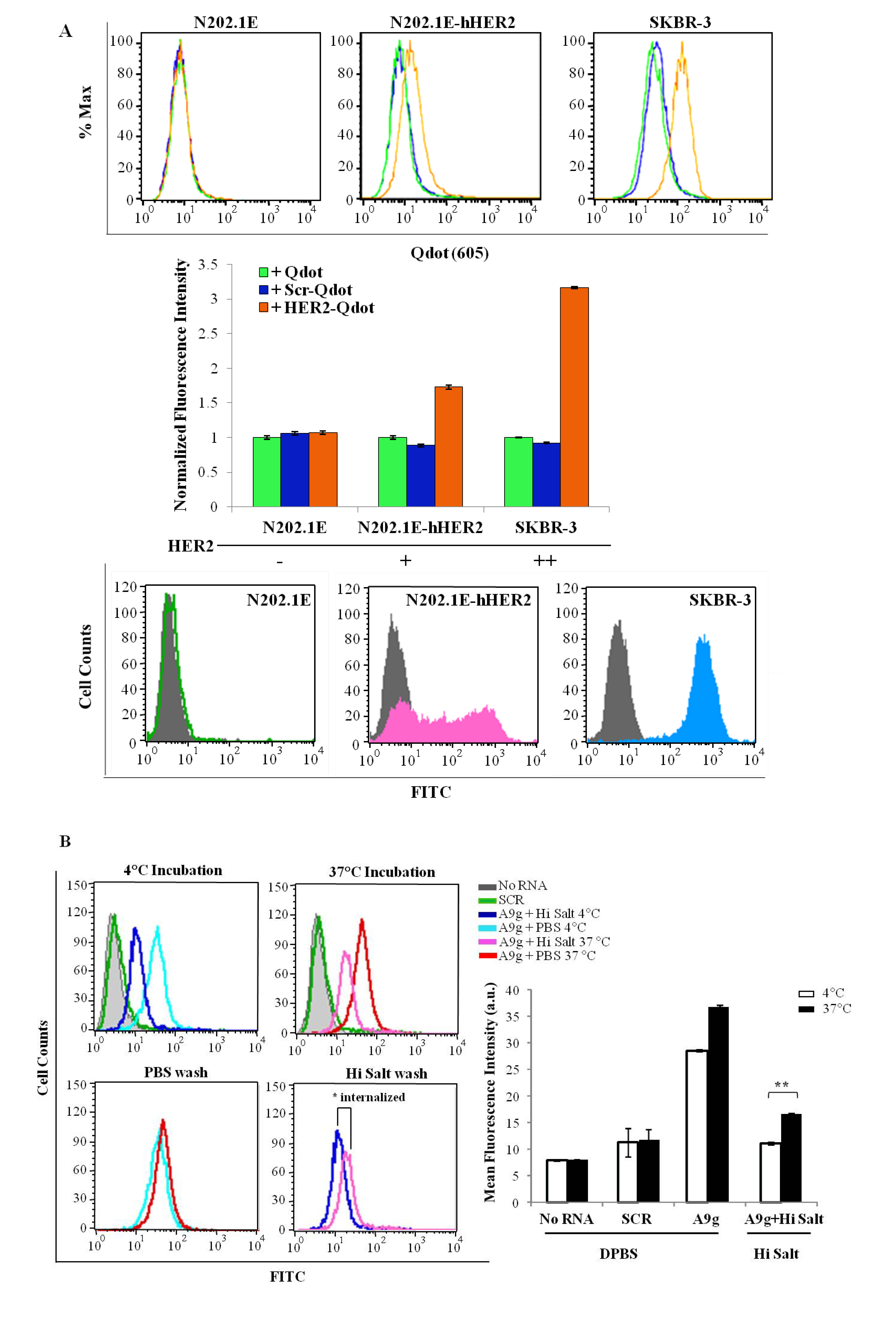
PSMA expression. PC3(PSMA+) and PC3(PSMA-) cells were grown in 60 mm cell culture dishes. On the day of the experiment cells were trypsinized (0.05% trypsin –EDTA), washed with DPBS and 2 × 105 cells/mL were incubated with 1 mL Block Buffer (4% FBS in DPBS) for 30 min at 25 °C. Next, cells were incubated for 30 min at 25 °C with a PE-conjugated monoclonal antibody against human PSMA (Cat# K0142-5) (1:100 dilution in Block) or Block containing a control antibody-PE. Then, cells were washed twice with 500 μL Block, fixed in 500 μL FIX (1% formaldehyde in DPBS) and analyzed by flow cytometry. PC3(PSMA-) cells served as a negative control cell line in this experiment.
HER2 expression. N202.1E, N202.1A, N202.1E-h HER2, and SK-BR-3 cells were cultured as described above. Cells were washed one time with DPBS, pelleted and resuspended in 100 μL of 1:200 dilution (in Block) of primary antibody against either human HER2-Ab-2 (NeoMarkers, Grand Island, NY, USA; cat # MS-229-P1), rat HER2 Ab-4 (Calbiochem, Fremont, CA, USA; cat# OP16) or 1:500 dilution (in Block) of isotype-specific control antibody. After 30 min incubation at 25 °C, cells were washed twice with 500 µL Block and incubated with 1:500 secondary antibody (goat anti-mouse Alexa Fluor488 IgG; cat# A11001) for 20 min at 25 °C in the dark. Cells were washed as detailed above and analyzed by flow cytometry.
3.3. RNA Aptamers
Unless noted otherwise, all RNA aptamer sequences were chemically synthesized with 2′ fluoropyrimidine (italics)–modified nucleotides obtained from TriLink Biotechnologies (San Diego, CA). Depending on the type of assay/application, aptamers were chemically synthesized with either 3'- or 5'-FAM, 5'-NH2 or 5'-biotin groups.
A9g aptamer
5'-GGGACCGAAAAAGACCUGACUUCUAUACUAAGUCUACGUUCCC-3'
A9g.6.aptamer
5'-GGGACCGAAAAAGACCUGGCUUCUAUACUAAGUCUACGUUCCC-3'
hHER2 aptamer
5'-AGCCGC GAGGGGAGGGAUAGGGUAGGGCGCGGCU-3'
E1 aptamer
5'‐GGGAGGACGAUGCGGUCCUGUCGUCUGUUCGUCCCCAGACGACUCGCCCGA‐3'
Scr aptamer
5'‐GGGAGGACGAUGCGGGACUAGCGAUCUGUUACGCACAGACGACUCGCCCGA‐3'
C4-3 aptamer
5'-GGGAGGACGAUGCGGUCGUAUUAUCCGCUGCACGCCAGACGACUCGCCCGA-3'
Scr2 aptamer
5'-GGGAGGACGAUGCGGUUUGGGGUUUUCCCGUGCCCCAGACGACUCGCCCGA-3'
3.4. Fluorescence Microscopy
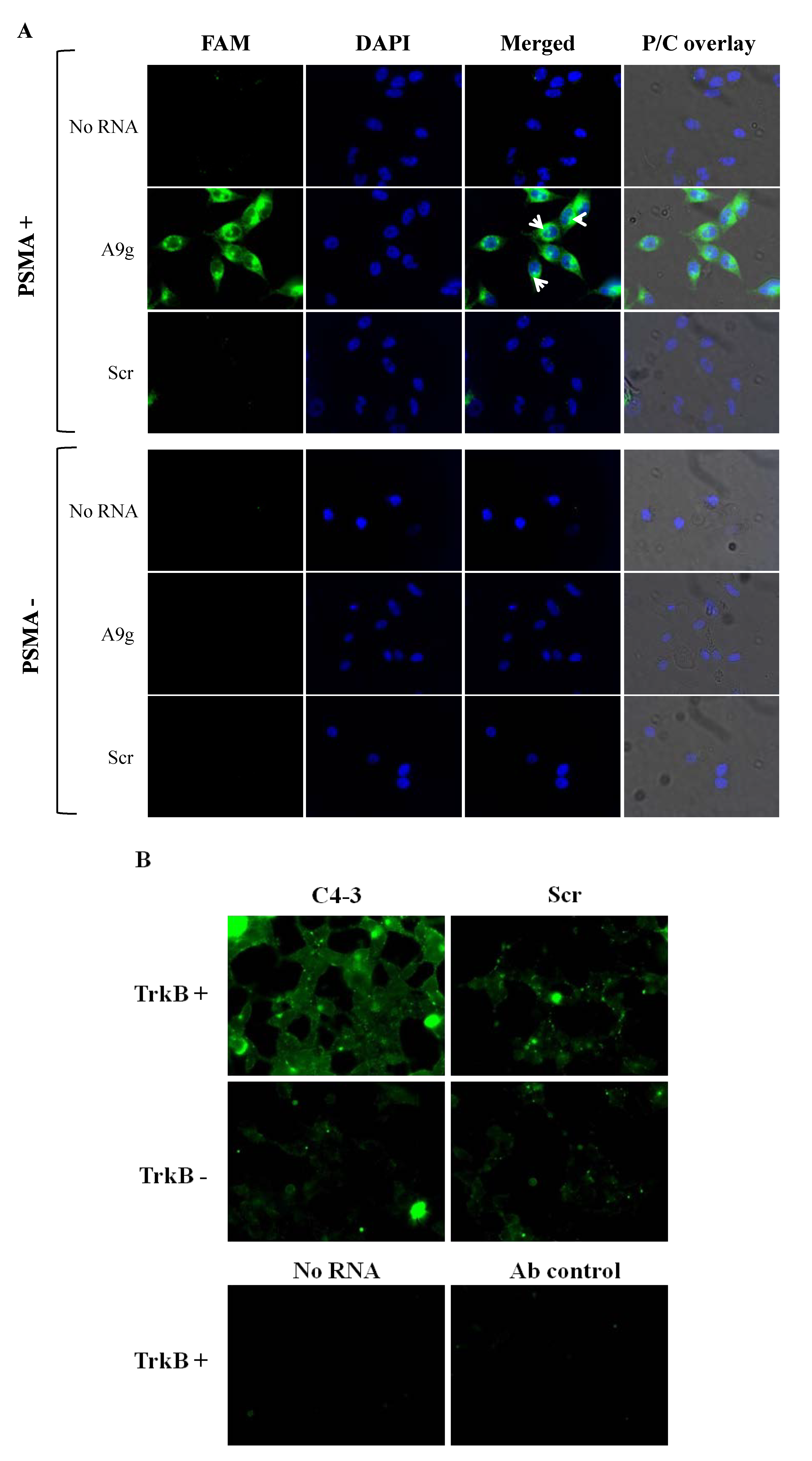
Direct method. For internalization experiments, PC3(PSMA+) and PC3(PSMA-) cells were grown on 35 mm glass-bottom MatTek culture dishes in DMEM/F12 media supplemented with 10% FBS. Cells were seeded at a density of 2 × 105 cells/mL in a volume of 500 μL one day prior to the experiment. On the day of the experiment, cells were washed with pre-warmed DPBS buffer and incubated for 30 min at 37 °C with 100 μL of 500 nM FAM-labeled aptamers (FAM on the 5'end) A9g (5'-GGGACCGAAAAAGACCUGACUUCUAUACUAAGUCUACGUUCCC -3') or control aptamer with a scrambled sequence: (5'‐GGGAGGACGAUGCGGGACUAGCGAUCUGUUACGCACAGAC GACUCGCCCGA‐3'). One hundred μL of DPBS alone was added to cells as no RNA control in these experiments. Next, cells were washed twice with ice-cold DPBS adjusted to 0.5 M NaCl (high salt wash) and 0.2 N acetic acid to remove unbound and surface bound RNAs. Subsequently, nuclei were stained with DAPI (1 g/mL) at room temperature for 15 min followed by washing three times with DPBS. For surface binding experiments (validation of the high salt wash experiments) cells were initially pre-conditioned by washing them with ice-cold DPBS. Subsequently, cold aptamer solutions or DPBS were incubated on cells at 4 °C for 10min. Nuclei were further stained as previously described and cells mounted for microscopy visualization.
Antibody amplification method. The antibody amplification protocol was carried out essentially as described previously [
18] with a few modifications. Briefly, HEK293 cells and TrkB-expressing HEK293 cells were grown on glass-bottom MatTek culture dishes in DMEM plus 10% FBS. Staining was performed as previously described [
18]. Cells were washed with DPBS pre-warmed at 37 °C following a 1 h incubation at 37 °C with FAM-labeled (FAM was on 3'end) C4-3 or control aptamer (100 nM RNA in 100µL) in DPBS. A non-specific, non-binding aptamer was used as a control in these experiment (Control aptamer: 5'-GGGA GGA
CGA
UG
CGG
UUUGGGG
UUUUCCCG
UG
CCCCAGA
CGA
CUC G
CCCGA-3'.
Following incubation with the RNA, cells were washed twice with ice-cold DPBS and then for 5 min with 0.5 M NaCl and 0.2 N acetic acid at 4 °C. Cells were then fixed for 20 min at room temperature with 4% formaldehyde, 4% sucrose in DPBS. The cells were permeabilized with 0.1% Triton X-100 in DPBS for 5 min. A block was performed by incubating cells with 5% goat serum plus 0.1% Triton X-100 in DPBS for 1 h at room temperature. The fluorescence signal was amplified by incubating with an anti-FITC antibody (rabbit anti-FITC, 1:1000 dilution; Invitrogen). The primary antibody incubation was followed by incubation with a fluorescent secondary antibody (goat anti-rabbit IgG, Alexa Fluor 488 conjugate, 1:1000 dilution; Invitrogen). Cells were mounted and coverslipped with a glycerol mounting medium.
All images were acquired with a 40X oil immersion objective, an Olympus IX71 inverted microscope, a cooled CCD camera and filters for FITC (excitation, 450–490 nm; emission, 515–565 nm). Data is representative of at least three captured images per condition. For the experiments with A9g aptamer, intracellular localization was further confirmed by overlaying the fluorescence images with the nuclear DAPI stain and subsequently, with the phase/contrast images using ImageJ 1.46 d software. The fluorescence images reported are representative of at least three captured images per dish/per condition. All microscopy experiments were repeated at least twice in different days.
3.5. Fluorescence Plate Reader
PC3(PSMA+), PC3(PSMA-) or N2021.A cells (expressing rat HER2) were grown to confluency in 150 mm culture dishes. Cells were then trypsinized and transferred to 1.5 mL Eppendorf tubes, in triplicate for each condition. Cells were centrifuged at 500 g for 3 min. Media was discarded and the cells were washed once with pre-warmed DPBS. For the binding experiments, cells were then fixed with 4% formaldehyde, 4% sucrose for 20 min at room temperature to inhibit internalization of aptamers. For the internalization experiments the fixation step was eliminated. Cells were further incubated with 100 µL of FAM-labeled chemically synthesized 2'fluoro RNAs (200 nM A9g, or 1 µM E1) for 1 h at 37 °C. After the RNA incubation, cells were subjected to either a DPBS wash to remove unbound aptamer, or a high salt wash (0.5 M NaCl) to remove unbound aptamers and aptamers bound to the cell membrane. For the high salt wash condition, cells were washed once briefly with both ice-cold DPBS and 0.5M NaCl, followed by a 5 min incubation with ice-cold 0.5M NaCl. Cells were further rinsed once with 1 mL DPBS and lysed using 400 µL of 0.1 N NaOH. Next, 100 µL of each lysate was loaded in triplicates onto a 96 well plate. Fluorescence was measured at 485/530 nm on an Analyst HT plate reader from LJL. At least two individual experiments per aptamer were performed using this assay.
3.6. Flow Cytometry
Binding of Qdot-aptamer conjugates. To prepare the Qdot-aptamer conjugates, 2.5 μL of 2 μM streptavidin-modified Quantum dots 605 (Invitrogen, cat# Q10001MP) were incubated with chemically synthesized, either 3'-biotin human HER2 aptamer (5'-AGCCGCGAGGGGAGGGAUAG GGUAGGGCGCGGCU-3') or 5'-biotin control aptamer (5'-GGGCCGAAAAAGACCUGACUUCU AUACUAAGUCUACGUCCC-3') (497.5 μL of 50 nM) for 30 min at 25 °C under gentle stirring. The final Qdot-aptamer conjugates were washed by centrifugal spin filtration, resuspended in DPBS, and then characterized using gel electrophoresis. Cells (N202.1E, N202.1E-hHER2 and SB-BR-3) were grown to confluency in 60 mm dishes in their respective media. In the day of the experiment, cells were harvested with 0.25% trypsin-EDTA (N202.1E and SK-BR-3) or 0.05% trypsin-EDTA (N202.1E-h HER2), washed once with media containing 10% FBS and further blocked in 10% FBS in DPBS for 30 min at room temperature. Next, cells were incubated with Qdot-hHER2 conjugate, Qdot-control aptamer conjugate or free Qdots for 45 min at 37 °C. After washing twice with 10% FBS in DPBS, cells were resuspended in DPBS and analyzed by flow cytometry using a Becton Dickinson LSR II equipped with a 633 nm laser for Qdot excitation.
Internalization of A9g-FAM. PC3(PSMA+) positive cells were cultured as described previously. Cells were trypsinized, washed twice with DPBS and 2 × 105 cells/mL aliquots prepared for internalization studies. Next, cells were incubated with 100 μL of 1 μM of either target aptamer (FAM-A9g), control aptamer (FAM-Scr) or DPBS for 30 min at either 4 °C (binding) or 37 °C (binding and internalization). Subsequently, cells were subjected to a DPBS wash or a high salt wash (0.5 M NaCl + 0.2 N acetic acid) to remove unbound and surface bound RNAs. The high salt wash was performed in two steps, as detailed above. This procedure ensures that majority of the surface bound sequences are removed, so that the detected signal is an accurate measure of specifically internalized RNAs. Finally, cells were suspended in DPBS and analyzed by flow cytometry.
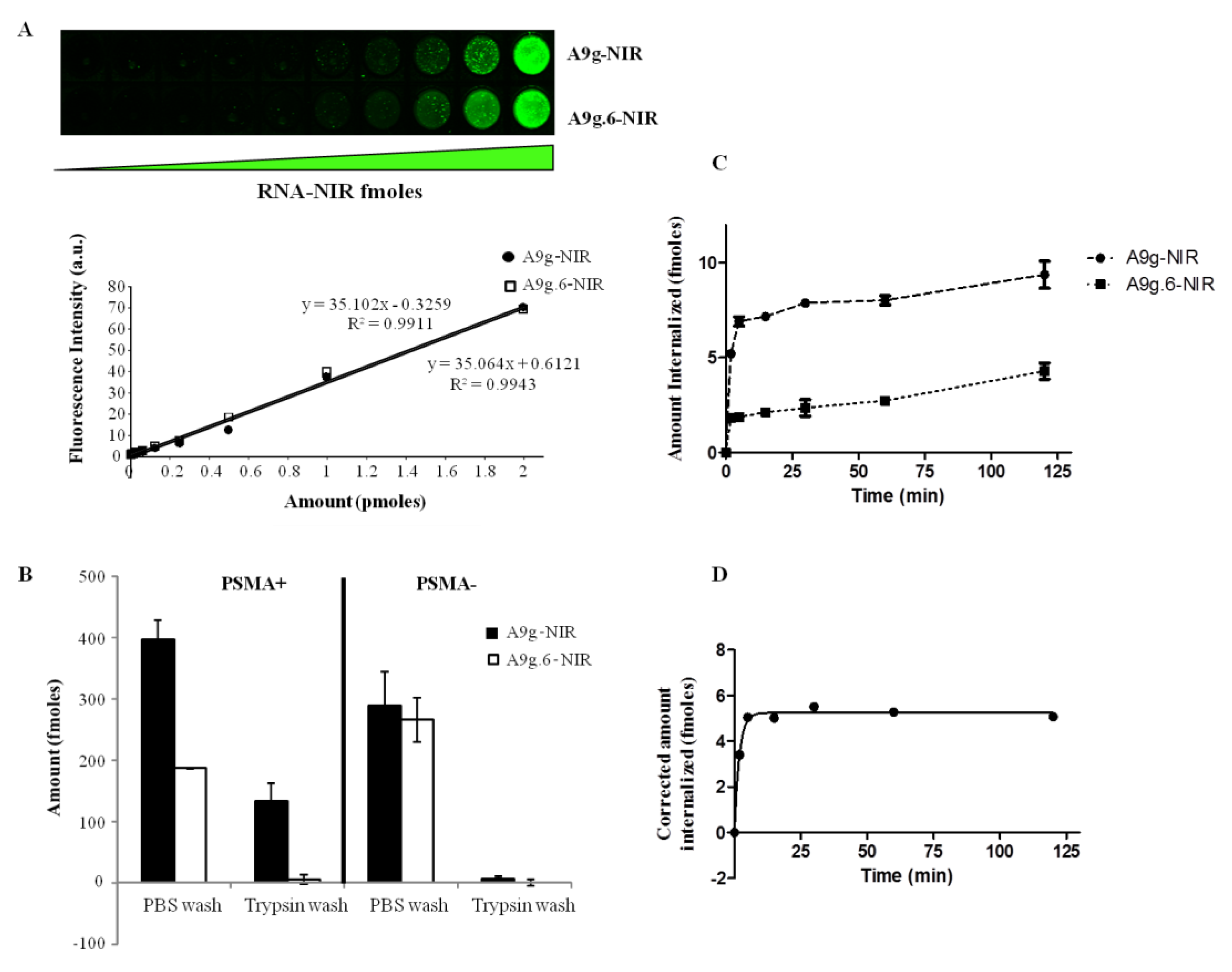
3.7. Quantification
Quantification of aptamer internalization was determined using IRDye800-labeled A9g (NIR-A9g). Specifically, 5x10
4 cells of either PC3(PSMA+) of PC3(PSMA-) cells were incubated with 100 μL of 10 nM of either target A9g-NIR or control (non-binding) A9g.6-NIR for 15 min at 37 °C. Cells were then washed with 500 μL of DPBS at room temperature or 100 μL of 0.25% trypsin-EDTA at 4°C for 30 min (to remove surface bound sequences). Cells that underwent trypsin wash were further washed two more times with 500 μL DPBS and finally, all samples were resuspended in 100 μL DPBS and transferred onto wells of a 96 well plate. Calibration curves for both, A9g-NIR and A9g.6-NIR, were constructed by measuring a series of twelve 1:2 dilutions starting from 10 nM for each aptamer. After sample loading, the plate was transferred to an Odyssey IR-Scanner (Li-COR, St. Lincoln, NE, USA) and scanned at 800 nm excitation using the supplied predefined settings for 96-well plate. For the time course studies, PC3 (PSMA+) cells were trypsinized with 0.05% trypsin-EDTA, washed with media containing 10% FBS and then resuspended in pre-warmed, serum free, DMEM/F12 media. Twenty thousand cells were incubated at 37 °C with A9g-NIR or A9g.6-NIR for the following time points: 120, 60, 30, 15, 5 min and no RNA sample serving as 0 time point. Internalization was halted by quickly transferring the samples to ice. Next, cells were incubated with 100 μL of 0.25% trypsin for 30 min at 4 °C to remove the surface bound RNAs, followed by a DPBS wash and then resuspended in 100 μL DPBS. Samples were loaded in 96 wells plate and scanned as described above. Aptamer uptake was time-dependent, showing an apparent exponential increase to a plateau value. Internalized aptamer data was fitted to a one-phase exponential association curve as described previously [
37] using GraphPad Prism Software (San Diego, CA, USA).
3.8. IRDye800CW Conjugated Aptamers(A9g-NIR and A9g.6-NIR)
A9g and A9g.6 aptamers, chemically synthesized with a 5'-NH2 group, were incubated with IRDye800 CW NHS ester (Li-Cor, cat# 929-70021) at a ratio of 1:40 (RNA:dye) for 2 h at 25 °C in 1 M potassium phosphate buffer (pH 9) under gentle stirring. Unreacted dye was removed by five successive spin filtrations. Aptamer labeling with IR dye was confirmed by ESI spectrometry and UV-Vis spectrophotometry. Functionality (binding) of the aptamer-NIR conjugates was tested by the PSMA NAALADase activity assay as described below.
3.9. PSMA NAALADase Activity Assay
The PSMA NAALADase activity assay was performed as previously described [
38] with minor modifications. Briefly, the final reaction volume was modified to 200 μL. Double-distilled H
2O (ddH
2O) was used for all solutions. The RNA aptamers were refolded in binding buffer (20 mM HEPES, 150 mM NaCl, 2 mM CaCl
2) at 1 μM concentration. Refolding was accomplished by heating at 65 °C for 10 min, followed by slow cooling to 37ºC for 10 min. Aptamers were concentrated through an Amicon 10,000 MW-cutoff spin filter (Cat# UFC801024). The remaining procedures were performed on ice. Ten microliters of refolded RNA in binding buffer was combined with 50 μL of 200 mM Tris buffer, pH 7.5, and 20 μL 10 mM CoCl
2. Recombinant PSMA was prepared by diluting 2 μg recombinant human PSMA (4234-ZN-010) from R&D Systems (Minneapolis, MN, USA) in 500 μL of 50 mM pH 7.5 Tris buffer. Ten microliters of the recombinant PSMA solution (40 ng PSMA) was added to the reaction mixture, and the reaction was incubated for 15 min at 37 °C to promote RNA-PSMA interaction. Ten microliters of a working solution containing 30 nM NAAG having a specific activity of 2.5 μCi/μL of [glutamate-3,4-3H]-NAAG from Perkin Elmer (Waltham, MA, USA; cat# NET1082250UC) was added to the reaction mixture. The reaction was allowed to proceed for 15 min, mixing once by gentle pipetting. The reaction was subsequently stopped by adding an equal volume (200 μL) of ice-cold 0.1M Na
2HPO
4 buffer. AG 1-X8 formate resin (200-400 mesh) from Bio-Rad (Hercules, CA, USA; cat# 140-1454) was used in columns to quantitate the [
3H]-glutamate reaction product. Before use, the columns were equilibrated with 2 mL of ddH
2O. Half of the final reaction volume (200 μL) was added to a column. The columns were eluted with 4 mL of 1 M formic acid and added to 10 mL of Bio-Safe II scintillation fluid (Research Products International Corp., Mt. Prospect, IL, USA). Activity was counted using a Beckman-Coulter liquid scintillation counter, and was normalized to the amount of activity obtained in the reaction with no RNA added.
3.10. RNA-RIP (Ribosome Inactivating Protein) Assay
Chemically synthesized, 5'-biotin RNA aptamers were reacted with a chemical conjugate of streptavidin and saporin toxin (Streptavidin-ZAP, Advanced Targeting Systems, San Diego, CA, USA; cat#IT-27) at room temperature for 30 min at a 1:1 molar ratio. The conjugates were characterized using gel electrophoresis. PSMA binding of aptamer-saporin conjugates was verified by PSMA NAALADase activity assay. PC3(PSMA-) and PC3(PSMA+) were seeded in 96 well plates (5 × 103 cells/well), grown overnight and then treated with different concentrations (1:2 dilution series) of A9g-saporin, A9g.6-saporin or FGF (basic fibroblast growth factor)-saporin (FGF-sap, Advanced Targeting Systems; cat #IT-38) for 72 h at 37 °C. The viability of treated cells was evaluated with Promega Cell Titer 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). Briefly, 20 μL of Cell Titer 96 AQueous One Solution was added to each well, including four wells containing only medium for background deduction. Cells were then incubated at 37 °C for 90 min. Absorbance at 490nm in each well was then determined using a Molecular Devices (Sunnyvale, CA, USA) ThermoMax microplate reader.
4. Conclusions and Discussion
In this study, we describe both qualitative and quantitative assays for characterizing the cell-type specific internalization ability of RNA aptamers for cell-targeted therapeutic applications. These assays include qualitative assays such as: (1) antibody amplification microscopy, (2) microplate-based fluorescence assay and (3) quantum-dot-based flow cytometry as well as a quantitative assay that we refer to as quantitative and ultrasensitive internalization method (“QUSIM”). In addition, we describe a novel way to monitor for cytoplasmic delivery using a ribosome inactivating protein-based assay (RNA-RIP assay). Since translocation of the siRNA to the cytoplasm is a crucial aspect of aptamer-targeted siRNA delivery, assays for monitoring translocation to the cytoplasm, like the RNA-RIP assay, are of importance. The next challenge will be to develop similar approaches capable of providing not only a qualitative, but also, a quantitative measure of the portion of the cell-associated siRNA that reaches the cytoplasm and is processed by the RNAi machinery.
Despite initial excitement and promise in clinical application of RNAi-based drugs, recent setbacks have tempered the excitement for these therapeutics and have highlighted the need for extensive efforts from both Industry and Academia to overcome these key hurdles. Effective cell-specific targeting and cytoplasmic delivery of siRNAs are two of the main limitations for the successful translation of this technology to humans. In recent years, RNA aptamers have emerged as potentially clinically useful and effective cell-targeting ligands for RNAi-based drugs [
39,
40,
41,
42,
43]. Importantly, these studies have also described a role for aptamers in facilitating delivery of siRNAs to the cytoplasm of target cells [
4,
5,
7,
9,
10,
11,
12,
13]. While, the precise mechanism for this is not well understood, it is generally accepted that efforts towards optimizing uptake into cells or escape from endosomes (two bottlenecks in intracellular delivery) would greatly enable the broad clinical application of this technology. Indeed, optimization of aptamer-mediated delivery of RNAi-based drugs will likely not only enhance efficacy and safety, but might also reduce the drug concentration and cost needed for effective therapy.
Methods for monitoring cellular uptake of cell-specific aptamers are the first step in optimizing reagents for targeted RNAi delivery. Many aptamer detection approaches involve adding multiple tags (fluorophores, biotin, etc.) to the body of the aptamer. One downside of these approaches is that they risk disrupting the aptamer structure. The alternative, which we prefer, involves end-labeling the aptamer with a single tag. However, in its simplest form (direct labeling: 1 tag/1 aptamer), this approach suffers from low sensitivity. To address this shortcoming, we have: (1) developed an antibody amplification method for the microscopy protocol, (2) coupled aptamers to quantum dots which are much brighter than conventional fluorophores (flow cytometry assay), (3) coupled aptamers to NIR dyes, which provide enhanced sensitivity compared to conventional fluorophores (“QUSIM” assay) and (4) coupled aptamers to a cellular toxin (saporin), which provides a sensitive cellular readout (RNA-RIP assay). As indicated above, the RNA-RIP assay not only provides a measure of cellular uptake but allows investigators to distinguish between those aptamers that internalize, but remain sequestered in endosomes and are eventually degraded by the lysosomal pathway, and those aptamers that escape the endosomes and are delivered to the cytoplasm of target cells where they engage into the RNAi pathway. This information may lead to further optimizations including attaching small molecules or combining with nanocarriers to enhance cellular uptake or endosomal escape.
In summary, the assays described herein provide powerful tools for assessing/confirming the internalization potential and sub-cellular localization of RNA aptamers within a target cell. Moreover, we show that the choice of assay to be performed depends largely on the nature of the aptamer sequence or that of its target receptor and suggest variations to protocols (see microscopy assay methodologies) for optimal sensitivity. Importantly, these efforts promise to expedite the development of RNA aptamer-based approaches for delivering siRNAs to the cytoplasm of target cells and possibly facilitate a more rapid translation of these therapeutics to humans.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




