
A Selenium Containing Inhibitor for the Treatment of Hepatocellular Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
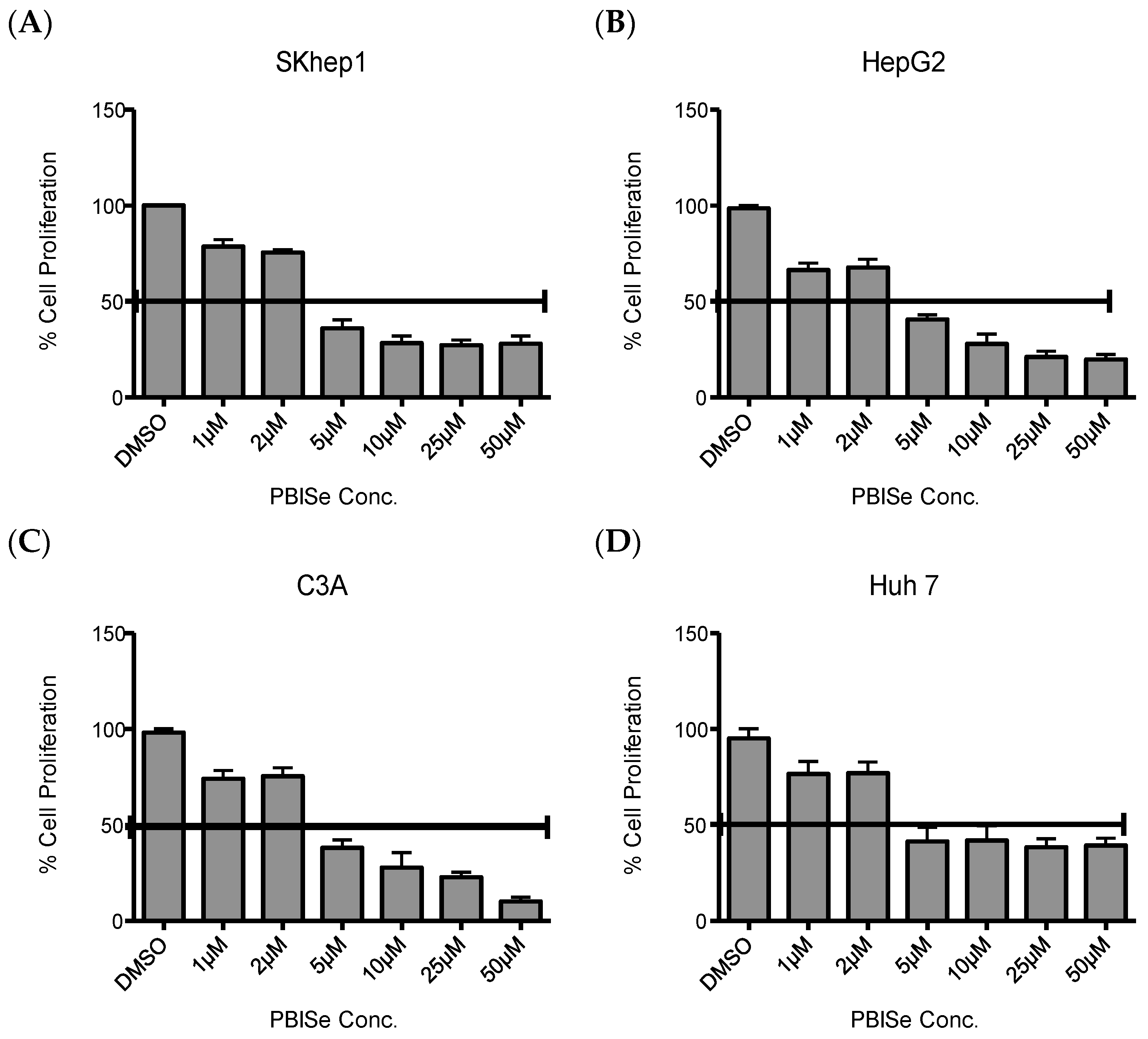
2.1. PBISe Inhibits Survival of Human HCC
2.2. PBISe Induces Apoptosis in Human HCC
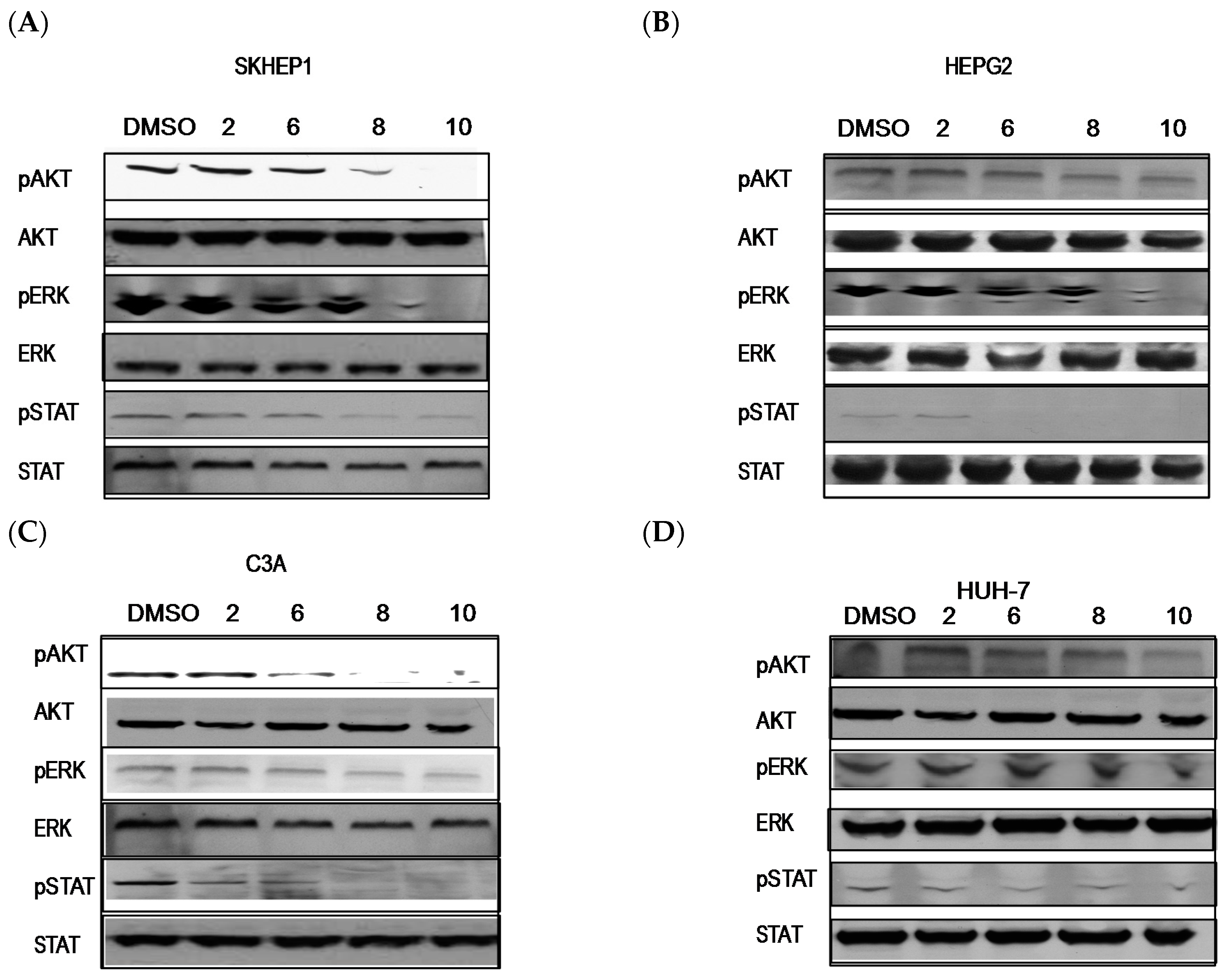
2.3. PBISe Inhibits MAPK, PI3K and STAT3 Pathways
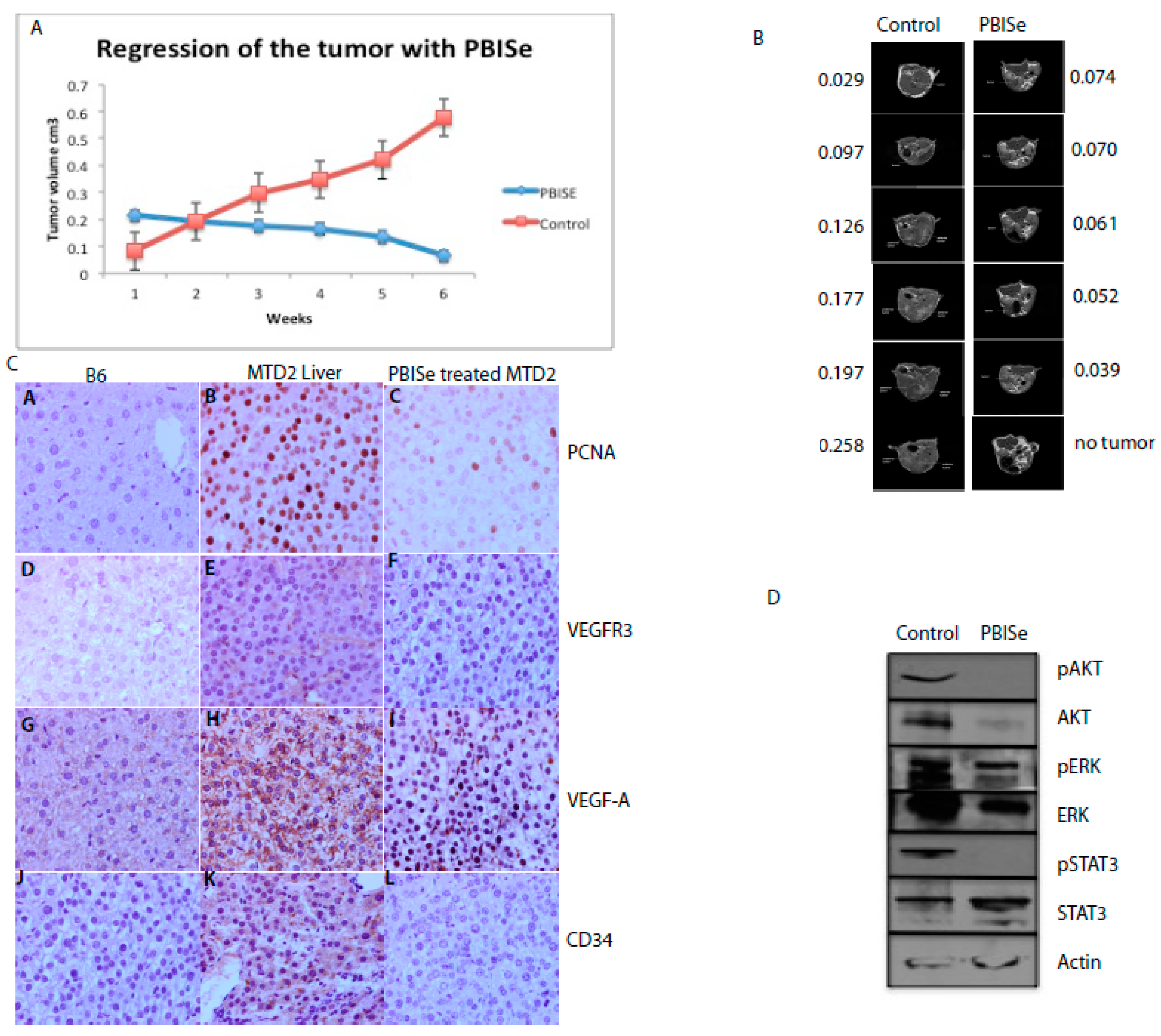
2.4. PBISe Inhibits In Vivo HCC Growth by Restricting Angiogenesis and Inducing Tumor Apoptosis
3. Discussion
4. Experimental Section
4.1. Synthesis of PBISe
4.2. Cell Lines and Culture Conditions
4.3. Cell Viability and Proliferation Assay
4.4. Apoptosis
4.6. Terminal Deoxynucleotide Transferase dUTP Nick End Labeling (TUNEL) Assay
4.7. Western Blot Analysis
4.8. Immunohistochemistry
4.9. In Vivo Tumor Model
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tong, M.J.; Hsien, C.; Song, J.J.; Kao, J.H.; Sun, H.E.; Hsu, L.; Han, S.H.; Durazo, F.A.; Saab, S.; Blatt, L.M. Factors associated with progression to hepatocellular carcinoma and to death from liver complications in patients with HBsAg-positive cirrhosis. Dig. Dis. Sci. 2009, 54, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; Seeff, L.B.; Morgan, T.R.; di Bisceglie, A.M.; Sterling, R.K.; Curto, T.M.; Everson, G.T.; Lindsay, K.L.; Lee, W.M.; Bonkovsky, H.L.; et al. Incidence of hepatocellular carcinoma and associated risk factors in hepatitis C-related advanced liver disease. Gastroenterology 2009, 36, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.H. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar]
- Lee, S.; Yoon, S.H.; Park, J.Y.; Kim, D.Y.; Ahn, S.H.; Han, K.H.; Choi, H.J. Sorafenib versus cytotoxic chemotherapy for patients with advanced hepatocellular carcinoma: A retrospective, single-institution study. Investig. New Drugs 2012, 30, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Guyton, K.Z.; Kensler, T.W. Prevention of liver cancer. Curr. Oncol. Rep. 2002, 4, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Egner, P.A.; Wang, J.B.; Zhu, Y.R.; Zhang, B.C.; Lu, P.X.; Chen, J.G.; Qian, G.S.; Kuang, S.Y.; Jackson, P.E.; et al. Chemoprevention of hepatocellular carcinoma in aflatoxin endemic areas. Gastroenterology 2004, 127, S310–S318. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Egner, P.A.; Wang, J.B.; Zhu, Y.R.; Zhang, B.C.; Qian, G.S.; Kuang, S.Y.; Gange, S.I.; Jacobson, L.P.; Munoz, A.; et al. Strategies for chemoprevention of liver cancer. Eur. J. Cancer Prev. 2002, 11 (Suppl. 2), S58–S64. [Google Scholar] [PubMed]
- Gurusamy, K. Trace element concentration in primary liver cancers—A systematic review. Biol. Trace Elem. Res. 2007, 118, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Seo, Y.R. Current issues of selenium in cancer chemoprevention. BioFactors 2010, 36, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hur, I.; Park, H.J.; Nam, J.; Park, G.B.; Kong, K.H.; Hwang, Y.M.; Kim, Y.S.; Cho, D.H.; Lee, W.J.; et al. Selenium inhibits metastasis of murine melanoma cells through the induction of cell cycle arrest and cell death. Immune Netw. 2009, 9, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Prabhu, K.S.; Das, A.; Mastro, A.M. Dietary selenium supplementationmodifies breast tumor growth and metastasis. Int. J. Cancer 2013, 133, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, V.; Plano, D.; Karelia, D.N.; Palop, J.A.; Amin, S.; Sanmartin, C.; Sharma, A.K. Novel seleno-and thio-urea derivatives with potent in vitro activities against several cancer cell lines. Eur. J. Med. Chem. 2016, 113, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Plano, D.; Karelia, D.N.; Pandey, M.K.; Spallholz, J.E.; Amin, S.; Sharma, A.K. Design, synthesis, and biological evaluation of novel selenium (Se-NSAID) molecules as anticancer agents. J. Med. Chem. 2016, 59, 1946–1959. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L. Selenium, selenoproteins and the thyroid gland: Interactions in health and disease. Nat. Rev. Endocrinol. 2011, 8, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Brozmanová, J.; Mániková, D.; Vlčková, V.; Chovanec, M. Selenium: A double-edged sword for defense and offence in cancer. Arch. Toxicol. 2010, 84, 919–938. [Google Scholar] [CrossRef] [PubMed]
- Muecke, R.; Schomburg, L.; Buentzel, J.; Kisters, K.; Micke, O. Selenium or no selenium—That is the question in tumor patients: A new controversy. Int. J. Mol. Sci. 2012, 13, 9649–9672. [Google Scholar] [CrossRef] [PubMed]
- Sanmartín, C.; Plano, D.; Palop, J.A. Selenium compounds and apoptotic modulation: A new perspective in cancer therapy. Mini Rev. Med. Chem. 2008, 8, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Nyandieka, H.S.; Wakhisi, J. The impact of vitamins A,C,E, and selenium compound on prevention of liver cancer in rats. East Afr. Med. J. 1993, 70, 151–153. [Google Scholar] [PubMed]
- Rao, C.V.; Indranie, C.; Simi, B.; Manning, P.T.; Connor, J.R.; Reddy, B.S. Chemopreventive properties of a selective inducible nitric oxide synthase inhibitor in colon carcinogenesis, administered alone or in combination with celecoxib, a selective cyclooxygenase-2 inhibitor. Cancer Res. 2002, 62, 165–170. [Google Scholar] [PubMed]
- Chen, T.; Nines, R.G.; Peschke, S.M.; Kresty, L.A.; Stoner, G.D. Chemopreventive effects of a selective nitric oxide synthase inhibitor on carcinogen-induced rat esophageal tumorigenesis. Cancer Res. 2004, 64, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Madhunapantula, S.V.; Desai, D.; Sharma, A.; Huh, S.J.; Amin, S.; Robertson, G.P. PBISe, a novel selenium-containing drug for the treatment of malignant melanoma. Mol. Cancer Ther. 2008, 7, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Madhunapantula, S.V.; Gowdahalli, K.; Sharma, A.; Chandagaludoreswamy, R.; El-Bayoumy, K.; Robertson, G.P.; Amin, S. Synthesis and characterization of a novel iNOS/Akt inhibitor Se,Se′-1,4-phenylenebis(1,2-ethanediyl)bisisoselenourea (PBISe)—Against colon cancer. Bioorg. Med. Chem. Lett. 2010, 20, 2038–2043. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Madhunapantula, S.V.; Desai, D.; Amin, S.; Robertson, G.P. Melanoma prevention using topical PBISe. Cancer Prev. Res. 2011, 4, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Bortner, J.; Desai, D.; Amin, S.; El-Bayoumy, K. The selenium analog of the chemopreventive compound S,S′-(1,4-phenylenebis[1,2-ethanediyl])bisisothiourea is a remarkable inducer of apoptosis and inhibitor of cell growth in human non-small cell lung cancer. Chem. Biol. Interact. 2009, 180, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. What is the role of thymidine phosphorylase in tumor angiogenesis. J. Natl. Cancer Inst. 1996, 88, 1091–1092. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.K.; Naora, H.; Yamanoi, A.; Ono, T.; Kohno, H.; Otani, H.; Nagasue, N. Requisite role of VEGF receptors in angiogenesis of hepatocellular carcinoma: A comparison with angiopoietin/Tie pathway. Anticancer Res. 2002, 22, 379–386. [Google Scholar] [PubMed]
- Poon, R.T.; Lau, C.P.; Cheung, S.T.; Yu, W.C.; Fan, S.T. Quantitative correlation of serum levels and tumor expression of vascular endothelial growth factor in patients with hepatocellular carcinoma. Cancer Res. 2003, 63, 3121–3126. [Google Scholar] [PubMed]
- Lian, Z.; Liu, J.; Wu, M.; Wang, H.Y.; Arbuthnot, P.; Kew, M.; Feitelson, M.A. Hepatitis B x antigen up-regulates vascular endothelial growth factor receptor 3 in hepatocarcinogenesis. Hepatology 2007, 45, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Jiang, W.; Ip, C.; Ganther, H.; Lu, J. Selenium-induced inhibition of angiogenesis in mammary cancer at chemopreventive levels of intake. Mol. Carcinpog. 1999, 26, 213–225. [Google Scholar] [CrossRef]
- Irmak, M.B.; Ince, G.; Ozturk, M.; Cetin-Atalay, R. Acquired tolerance of hepatocellular carcinoma cells to selenium deficiency: A selective survival mechanism? Cancer Res. 2003, 63, 6707–6715. [Google Scholar] [PubMed]
- Peters, U.; Takata, Y. Selenium and the prevention of prostate and colorectal cancer. Mol. Nutr. Food Res. 2008, 52, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Combs, G.F., Jr. Chemopreventive mechanisms of selenium. Med. Klin. 1999, 94, 18–24. [Google Scholar] [CrossRef]
- Costantini, S.; Lepore, M.G.; Castello, G.; Colonna, G. Has selenium a chemopreventive effect on 468 hepatocellular carcinoma? Mini Rev. Med. Chem. 2011, 11, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, R.C.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 2011, 192–222. [Google Scholar] [CrossRef]
- Schmitz, K.J.; Wohlschlaeger, J.; Lang, H.; Sotiropoulos, G.C.; Malago, M.; Steveling, K.; Reis, H.; Cicinnati, V.R.; Schmid, K.W.; Baba, H.A. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J. Hepatol. 2008, 48, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Sakamoto, M.; Yamasaki, S.; Todo, S.; Hirohashi, S. Akt phosphorylation is a risk factor for early disease recurrence and poor prognosis in hepatocellular carcinoma. Cancer 2005, 103, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Yim, S.Y.; Chae, K.R.; Shim, S.B.; Hong, J.T.; Park, J.Y.; Lee, C.Y.; Son, H.J.; Sheen, Y.Y.; Hwang, D.Y. ERK activation induced by selenium treatment significantly downregulates β/γ-secretase activity and Tau phosphorylation in the transgenic rat overexpressing human selenoprotein M. Int. J. Mol. Med. 2009, 24, 91–96. [Google Scholar] [PubMed]
- Chen, T.; Wong, Y.S.; Zheng, W. Induction of G1 cell cycle arrest and mitochondria-mediated apoptosis in MCF-7 human breast carcinoma cells by selenium-enriched Spirulina extract. Biomed. Pharmacother. 2009, 63, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Wang, Z.; Ganther, H.; Lu, J. Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 2001, 61, 3062–3070. [Google Scholar] [PubMed]
- Xu, J.; Yang, F.; An, X.; Hu, Q. Anticarcinogenic activity of selenium-enriched green tea extracts in vivo. J. Agric. Food Chem. 2007, 55, 5349–5353. [Google Scholar] [CrossRef] [PubMed]
- Popova, N.V. Perinatal selenium exposure decreases spontaneous liver tumorogenesis in CBA mice. Cancer Lett. 2002, 179, 39–42. [Google Scholar] [CrossRef]
- Novoselov, S.V.; Calvisi, D.F.; Labunskyy, V.M.; Factor, V.M.; Carlson, B.A.; Fomenko, D.E.; Moustafa, M.E.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein deficiency and high levels of selenium compounds can effectively inhibit hepatocarcinogenesis in transgenic mice. Oncogene 2005, 24, 8003–8011. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, M.; Mizrahi, L.; Pappo, O.; Klopstock, N.; Olam, D.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Domany, E.; Galun, E.; et al. Molecular mechanisms of liver carcinogenesis in the mdr2-knockout mice. Mol. Cancer Res. 2007, 5, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; O’Connor, L.; Rossman, T.G.; Block, E. Pro-angiogenesis action of arsenic and its reversal by selenium-derived compounds. Carcinogenesis 2007, 28, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Avella, D.M.; Li, G.; Schell, T.D.; Liu, D.; Zhang, S.S.; Lou, X.; Berg, A.; Kimchi, E.T.; Tagaram, H.R.; Yang, Q.; et al. Regression of established hepatocellular carcinoma is induced by chemoimmunotherapy in an orthotopic murine model. Hepatology 2012, 55, 141–152. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2004. [Google Scholar]
- Cedergreen, N.; Ritz, C.; Streibig, J.C. Improved empirical models describing hormesis. Environ. Toxicol. Chem. 2005, 24, 3166–3172. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tagaram, H.R.S.; Desai, D.; Li, G.; Liu, D.; Rountree, C.B.; Gowda, K.; Berg, A.; Amin, S.; Staveley-O’Carroll, K.F.; Kimchi, E.T. A Selenium Containing Inhibitor for the Treatment of Hepatocellular Cancer. Pharmaceuticals 2016, 9, 18. https://doi.org/10.3390/ph9020018
Tagaram HRS, Desai D, Li G, Liu D, Rountree CB, Gowda K, Berg A, Amin S, Staveley-O’Carroll KF, Kimchi ET. A Selenium Containing Inhibitor for the Treatment of Hepatocellular Cancer. Pharmaceuticals. 2016; 9(2):18. https://doi.org/10.3390/ph9020018
Chicago/Turabian StyleTagaram, Hephzibah Rani S., Dhimant Desai, Guangfu Li, Dai Liu, C. Bart Rountree, Kavitha Gowda, Arthur Berg, Shantu Amin, Kevin F. Staveley-O’Carroll, and Eric T. Kimchi. 2016. "A Selenium Containing Inhibitor for the Treatment of Hepatocellular Cancer" Pharmaceuticals 9, no. 2: 18. https://doi.org/10.3390/ph9020018
APA StyleTagaram, H. R. S., Desai, D., Li, G., Liu, D., Rountree, C. B., Gowda, K., Berg, A., Amin, S., Staveley-O’Carroll, K. F., & Kimchi, E. T. (2016). A Selenium Containing Inhibitor for the Treatment of Hepatocellular Cancer. Pharmaceuticals, 9(2), 18. https://doi.org/10.3390/ph9020018