
Molecular Structure-Based Screening of the Constituents of Calotropis procera Identifies Potential Inhibitors of Diabetes Mellitus Target Alpha Glucosidase

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preprocessing of Target Structure
2.2. Molecular Docking of Compounds against Alpha Glucosidase
2.3. Mechanism of Binding Characterization
2.4. Validation of Docking Protocol
2.5. Prediction of Biological Activity of Compounds
2.6. Molecular Dynamics Simulations of Protein-Ligand Complexes
2.7. MM-PBSA Calculations of Receptor-Ligand Complex
2.8. Structural Exploration of Potential Leads
3. Results and Discussion
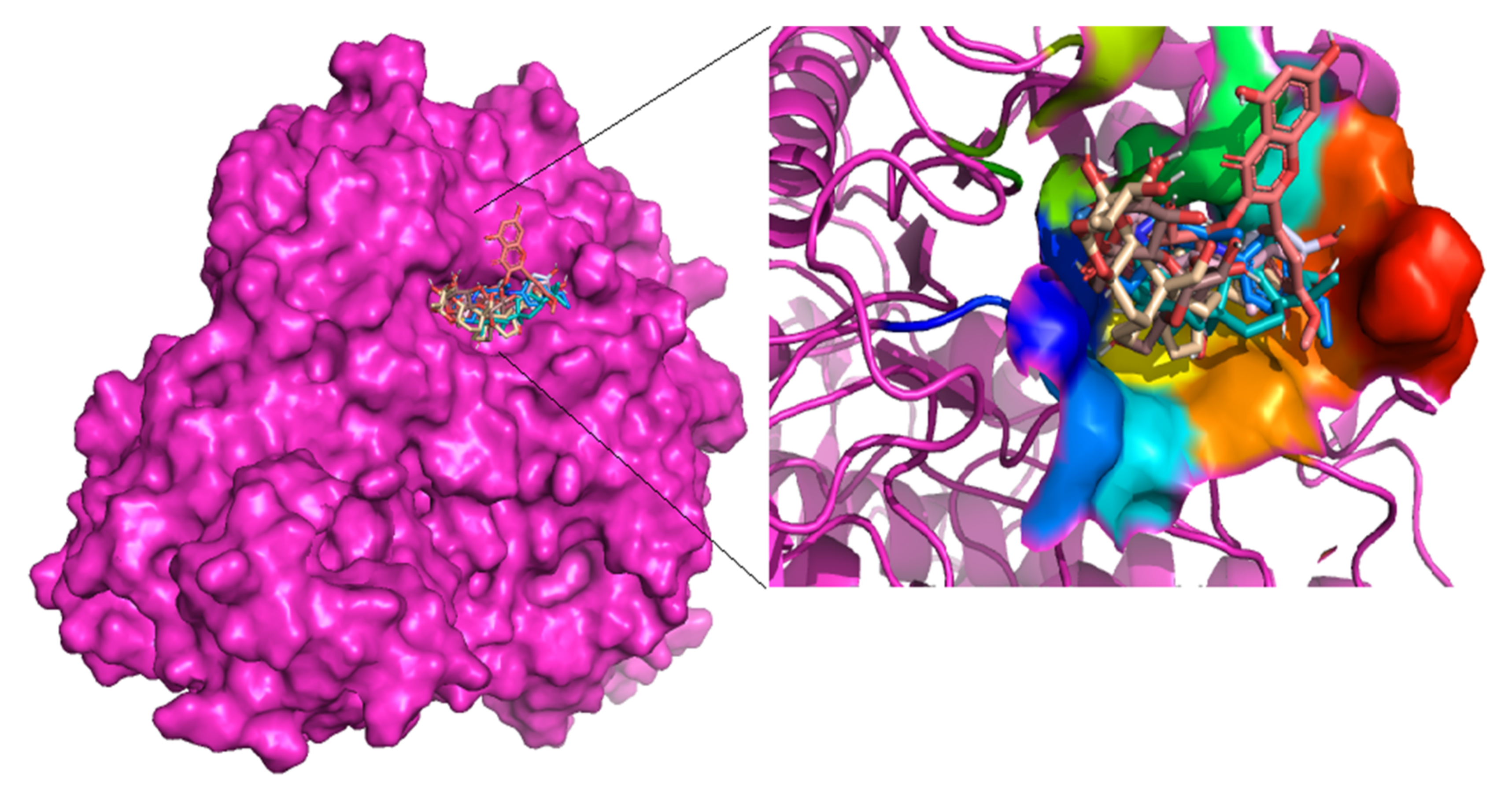
3.1. Preprocessing of Alpha Glucosidase as a Target Structure
3.2. Molecular Docking against Alpha Glucosidase as a Target Structure
3.3. Comparison of Binding Energies of Selected Compounds of Calotropis procera and Known Inhibitors
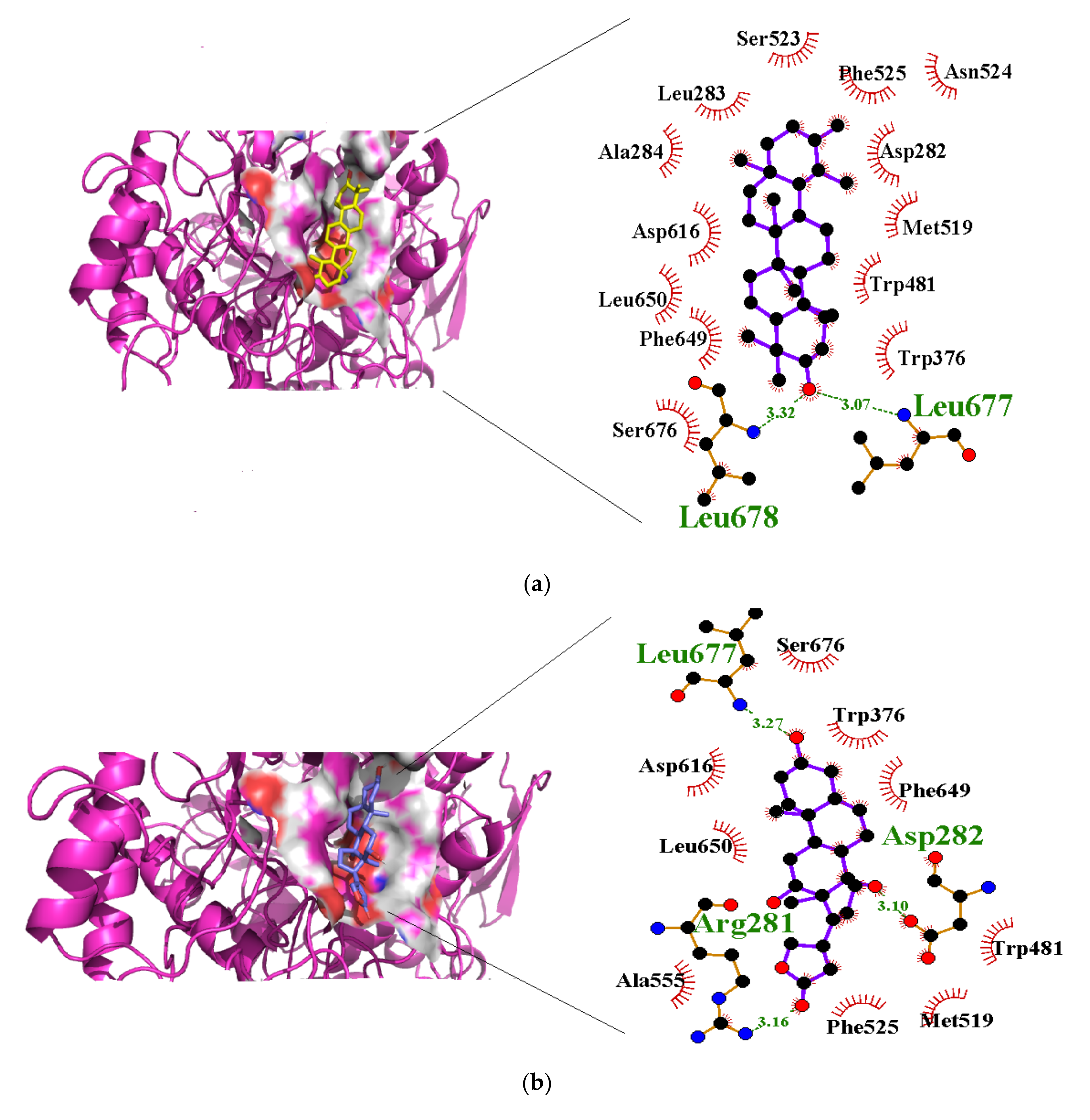
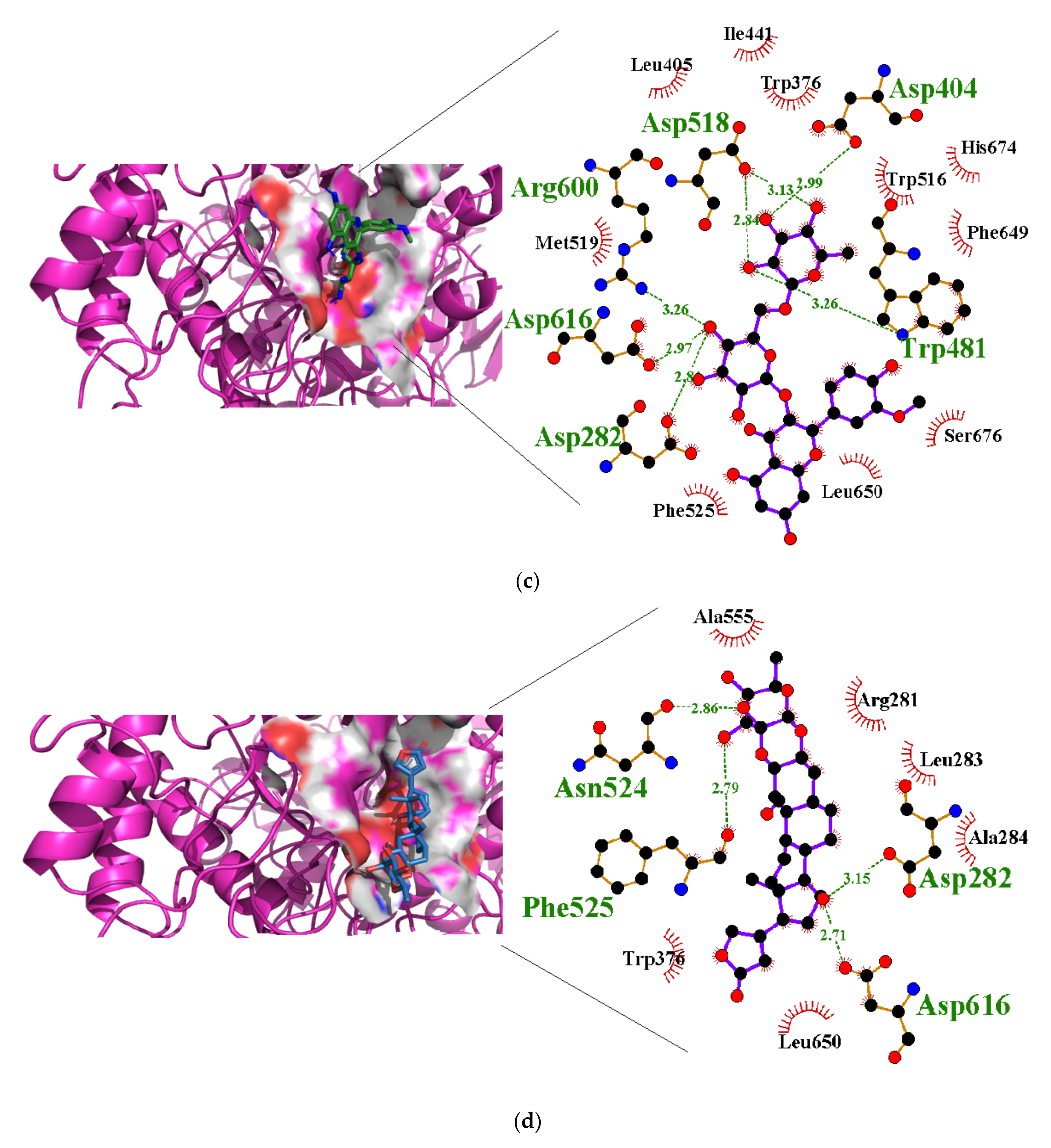
3.4. Molecular Interactions with Alpha Glucosidase
3.5. Validation of Docking Protocol
3.6. Prediction of Antidiabetic Activity of Selected Compounds
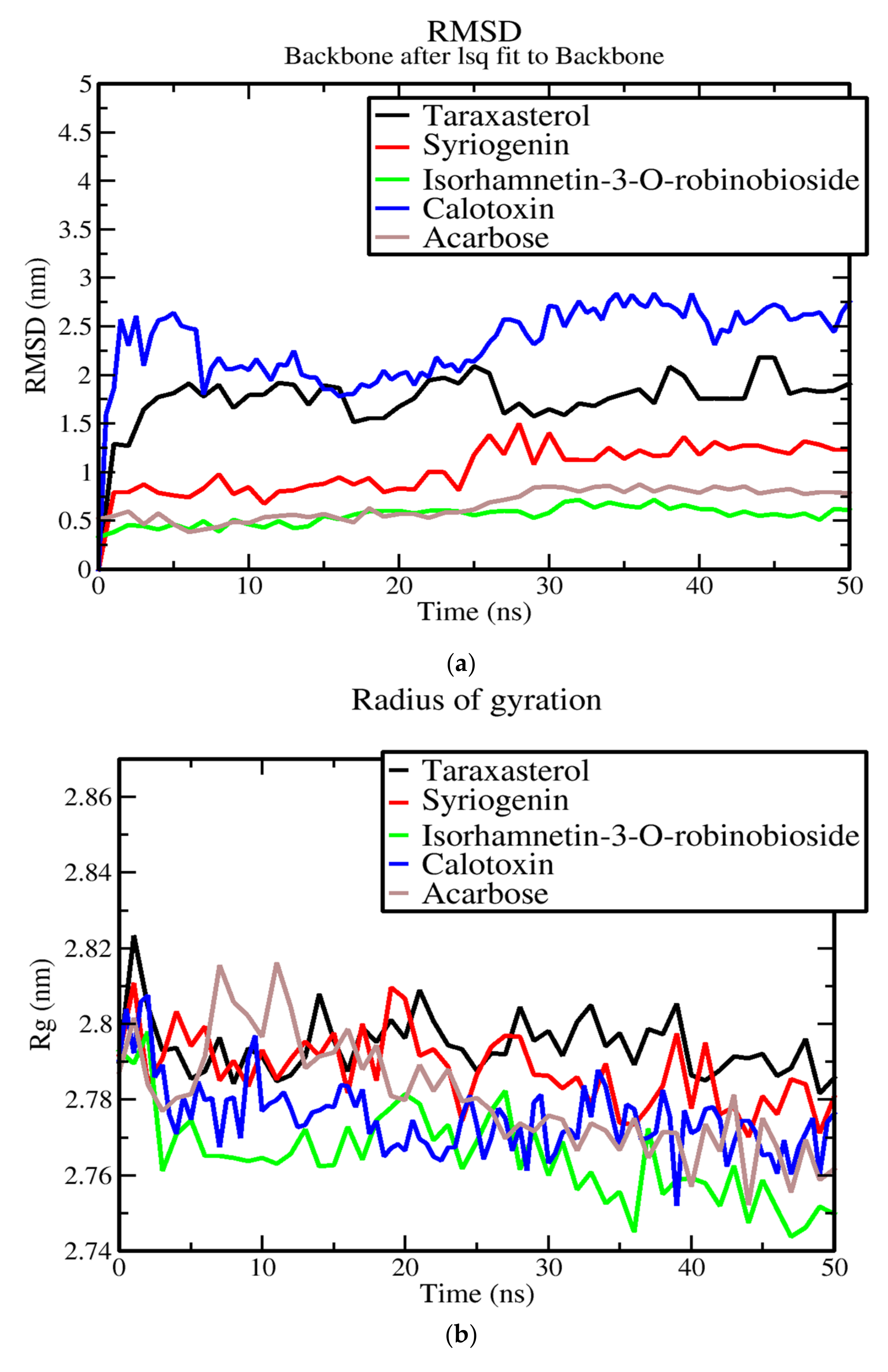
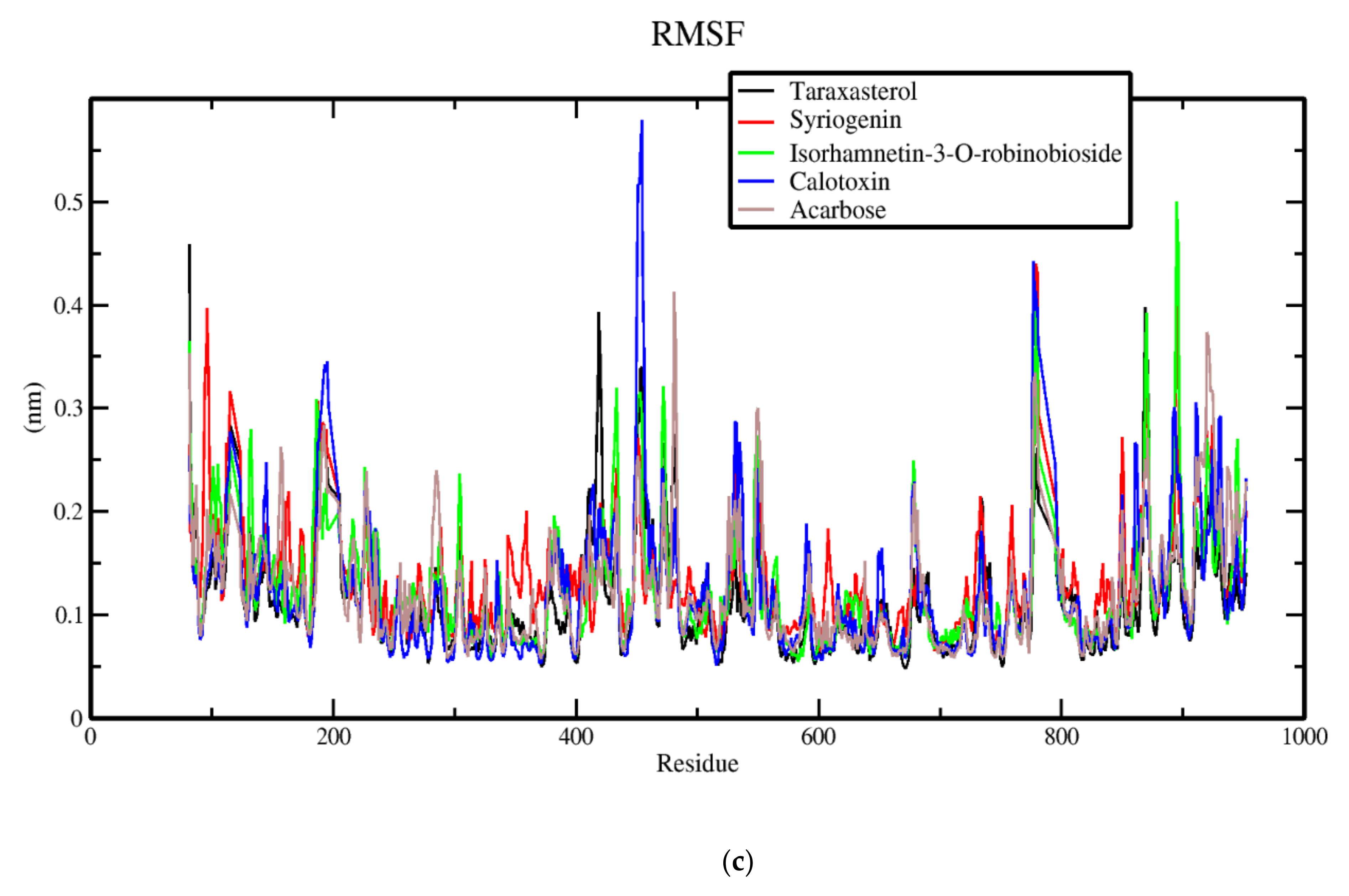
3.7. Molecular Dynamics of Protein-Ligand Complex of Potential Leads
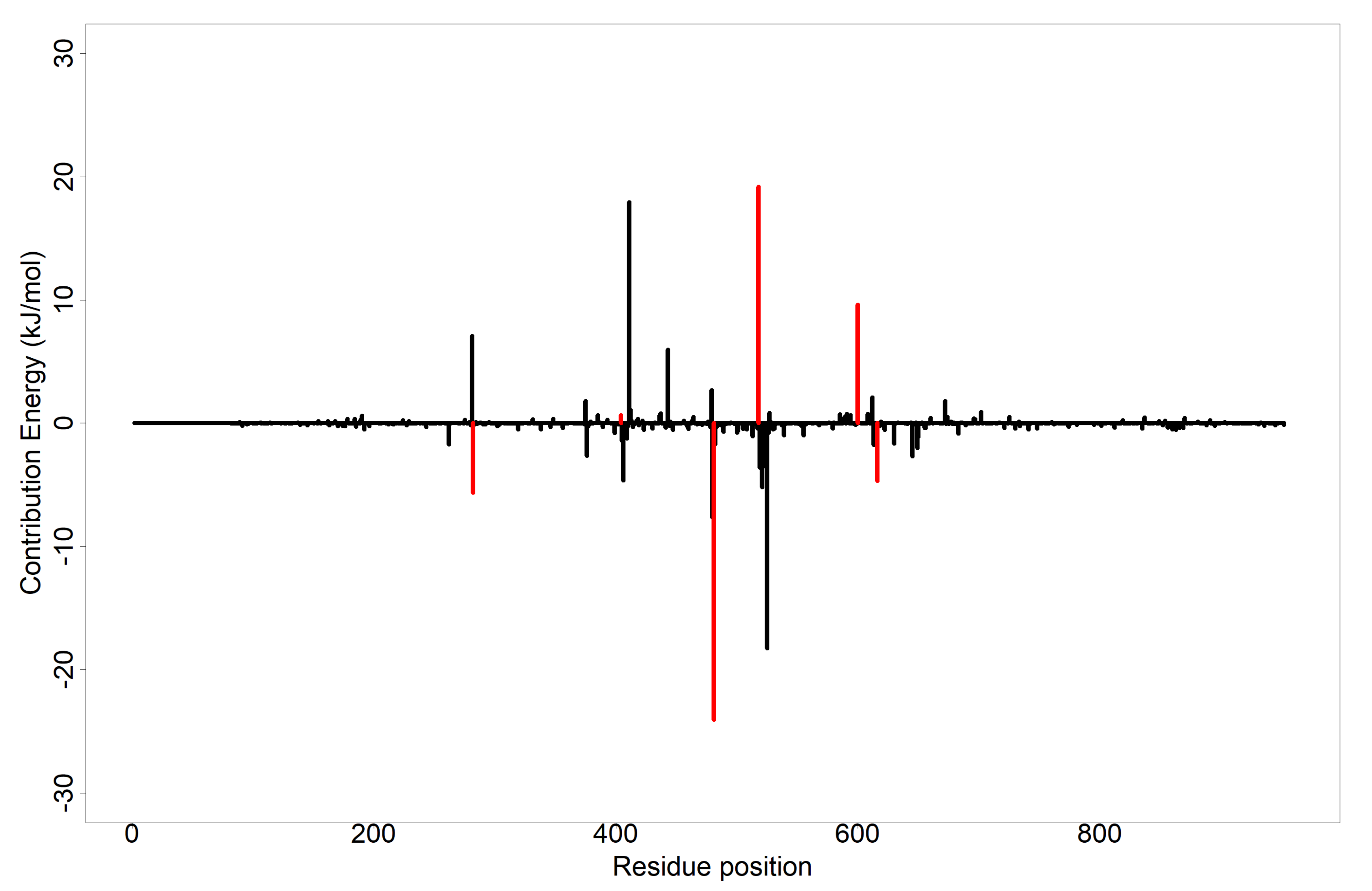
3.8. Evaluation of Putative Leads Using MM-PBSA Approach
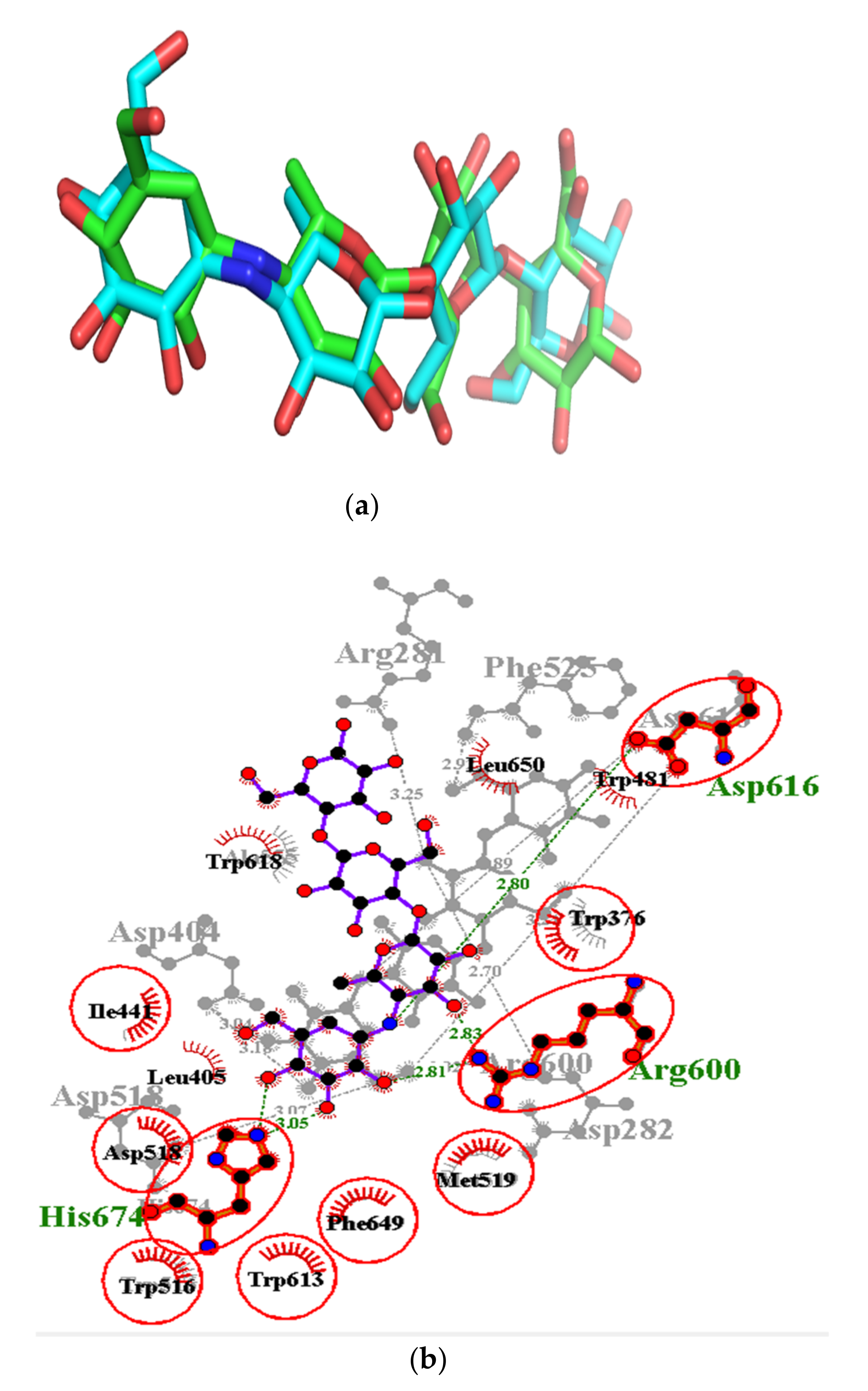
3.9. Exploring Possible Structural Similarity of Predicted Leads
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sen, S.; Chakraborty, R. EDITORIAL (Thematic issue: Treatment and Diagnosis of Diabetes Mellitus and Its Complication: Advanced Approaches). Mini-Reviews Med. Chem. 2015, 15, 1132–1133. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Diagnosis and classification of diabetes mellitus, ADA Clinical Practice Recommendations. Diabetes Care 2013, 36 (Suppl. S1), S67–S74. [CrossRef] [Green Version]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Publ. Gr. 2017, 14, 88–98. [Google Scholar] [CrossRef]
- Harreiter, J.; Roden, M. Diabetes mellitus—Definition, classification, diagnosis, screening and prevention (Update 2019). Wien. Klin. Wochenschr. 2019, 131, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Guasch-ferré, M.; Merino, J.; Sun, Q.; Fitó, M.; Salas-salvadó, J. Review Article Dietary Polyphenols, Mediterranean Diet, Prediabetes, and Type 2 Diabetes: A Narrative Review of the Evidence. Oxid. Med. Cell. Longev. 2017, 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.; Vijayan, R. Identification of novel aldose reductase inhibitors from spices: A molecular docking and simulation study. PLoS ONE 2015, 10, e0138186. [Google Scholar] [CrossRef]
- Dey, B.; Mitra, A.; Katakam, P.; Singla, R.K. Exploration of natural enzyme inhibitors with hypoglycemic potentials amongst Eucalyptus Spp. by in vitro assays. World J. Diabetes 2014, 5, 209. [Google Scholar] [CrossRef]
- William, J.; John, P.; Mumtaz, M.W.; Ch, A.R.; Adnan, A.; Mukhtar, H.; Sharif, S.; Raza, S.A.; Akhtar, M.T. Antioxidant activity, α-glucosidase inhibition and phytochemical profiling of Hyophorbe lagenicaulis leaf extracts. PeerJ 2019, 7, e7022. [Google Scholar] [CrossRef] [Green Version]
- Azam, S.S.; Uddin, R.; Wadood, A. Structure and dynamics of alpha-glucosidase through molecular dynamics simulation studies. J. Mol. Liq. 2012, 174, 58–62. [Google Scholar] [CrossRef]
- Kalra, S.; Osonoi, T. Alpha-glucosidase inhibitor. Nihon Rinsho. 2015, 73, 390–394. [Google Scholar] [CrossRef]
- Laar, F. Alpha-glucosidase inhibitors in the early treatment of type 2 diabetes. Vasc. Health Risk Manag. 2008, 4, 1189–1195. [Google Scholar] [CrossRef] [Green Version]
- Marella, S.; Maddirela, D.R.; Kumar, E.G.T.V.; Tilak, T.K.; Badri, K.R.; Chippada, A. Mcy protein, a potential antidiabetic agent: Evaluation of carbohydrate metabolic enzymes and antioxidant status. Int. J. Biol. Macromol. 2016, 86, 481–488. [Google Scholar] [CrossRef]
- Chandran, M. Diabetes Drug Effects on the Skeleton. Calcif. Tissue Int. 2017, 100, 133–149. [Google Scholar] [CrossRef]
- Xu, L.; Li, Y.; Dai, Y.; Peng, J. Natural products for the treatment of type 2 diabetes mellitus: Pharmacology and mechanisms. Pharmacol. Res. 2018, 130, 451–465. [Google Scholar] [CrossRef]
- Necyk, C.; Zubach-Cassano, L. Natural Health Products and Diabetes: A Practical Review. Can. J. Diabetes 2017, 41, 642–647. [Google Scholar] [CrossRef]
- Kalhotra, P.; Chittepu, V.C.S.R.; Osorio-Revilla, G.; Gallardo-Velázquez, T. Structure–activity relationship and molecular docking of natural product library reveal chrysin as a novel dipeptidyl peptidase-4 (DPP-4) inhibitor: An integrated in silico and in vitro study. Molecules 2018, 23, 1368. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Chaphalkar, S.R.; Pratishthan, V. Anti-Diabetic Activity of Calotropis Gigantea in Human Whole Blood Journal of Disease and Global Anti-Diabetic Activity of Calotropis Gigantea in Human Whole Blood. J. Dis. Glob. Health 2016, 107–112. [Google Scholar]
- Kazeem, M.I.; Mayaki, A.M.; Ogungbe, B.F.; Ojekale, A.B. In-vitro studies on Calotropis procera leaf extracts as inhibitors of key enzymes linked to diabetes mellitus. Iran. J. Pharm. Res. 2016, 15, 37–44. [Google Scholar] [CrossRef]
- Al-snafi, A.E. The Constituents and Pharmacological Properties of Calotropis Procera-An Overview. Int. J. Pharm. Rev. Res. 2015, 5, 259–275. [Google Scholar]
- Bairagi, S. Pharmacology of Natural Products: An recent approach on Calotropis gigantea and Calotropis procera. Ars Pharm. 2018, 59, 37–44. [Google Scholar] [CrossRef]
- Mohamed, M.A.; Hamed, M.M.; Ahmed, W.S.; Abdou, A.M. Antioxidant and cytotoxic flavonols from Calotropis procera. Zeitschrift fur Naturforsch.-Sect. C J. Biosci. 2011, 66, 547–554. [Google Scholar] [CrossRef] [Green Version]
- Morsy, N.M.; Sherif, E.A. Al Phytochemical analysis of Calotropis procera with antimicrobial activity investigation. Main Gr. Chem. 2016, 11, 267–273. [Google Scholar] [CrossRef]
- Ahmad, M.B.; Gwarzo, M.Y.; Anwar, S. Antioxidative and anti-hyperglycaemic effect of calotropis procera in alloxan induced diabetic rats. J. Med. Plants Res. 2016, 10, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, V.; Ajay, S.S. Antihyperglycemic and antihyperlipidaemic activities of root extracts of Calotropis procera (Ait.) R. Br on streptozotocin induced diabetic rats. Jordan J. Biol. Sci. 2009, 2, 177–180. [Google Scholar]
- Neto, M.C.L.; de Vasconcelos, C.F.B.; Thijan, V.N.; Caldas, G.F.R.; Araújo, A.V.; Costa-Silva, J.H.; Amorim, E.L.C.; Ferreira, F.; de Oliveira, A.F.M.; Wanderley, A.G. Evaluation of antihyperglycaemic activity of Calotropis procera leaves extract on streptozotocin-induced diabetes in Wistar rats. Brazilian J. Pharmacogn. 2013, 23, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Baira, S.M.; Sigalapalli, D.K.; Bathini, N.B.; Srinivas, R.; Talluri, M.V.N.K. LC/QTOF/MS/MS characterization, molecular docking and in silico toxicity prediction studies on degradation products of anagliptin. J. Pharm. Biomed. Anal. 2018, 159, 92–99. [Google Scholar] [CrossRef]
- Jia, Y.; Ma, Y.; Cheng, G.; Zhang, Y.; Cai, S. Comparative Study of Dietary Flavonoids with Different Structures as α-Glucosidase Inhibitors and Insulin Sensitizers. J. Agric. Food Chem. 2019, 67, 10521–10533. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Muhammad, I.; Nayab, G.-E.; Khan, H.; Aschner, M.; Filosa, R.; Daglia, M. Molecular Docking of Isolated Alkaloids for Possible α-Glucosidase Inhibition. Biomolecules 2019, 9, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Singh, V.K.; Singh, A.K. Molecular docking analysis of candidate compounds derived from medicinal plants with type 2 diabetes mellitus targets. Bioinformation 2019, 15, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benalla, W.; Bellahcen, S.; Bnouham, M. Antidiabetic Medicinal Plants as a Source of Alpha Glucosidase Inhibitors. Curr. Diabetes Rev. 2010, 6, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Molecular Mechanism in α-Glucosidase and Glucoamylase. Biosci. Biotechnol. Biochem. 1997, 61, 1233–1239. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, S.; Lee, H.S.; Kim, B.K.; Ohuchi, K.; Shin, K.H. Inhibitory effects of isorhamnetin-3-O-β-D-glucoside from salicornia herbacea on rat lens aldose reductase and sorbitol accumulation in streptozotocin-induced diabetic rat tissues. Biol. Pharm. Bull. 2005, 28, 916–918. [Google Scholar] [CrossRef] [Green Version]
- Hakamata, W.; Kurihara, M.; Okuda, H.; Nishio, T.; Oku, T. Design and Screening Strategies for α-Glucosidase Inhibitors Based on Enzymological Information. Curr. Top. Med. Chem. 2009, 9, 3–12. [Google Scholar] [CrossRef]
- Tundis, R.; Loizzo, M.R.; Menichini, F. Natural Products as α-Amylase and α-Glucosidase Inhibitors and their Hypoglycaemic Potential in the Treatment of Diabetes: An Update. Mini-Rev. Med. Chem. 2010, 10, 315–331. [Google Scholar] [CrossRef]
- Yue, L.M.; Lee, J.; Zheng, L.; Park, Y.D.; Ye, Z.M.; Yang, J.M. Computational prediction integrating the inhibition kinetics of gallotannin on α-glucosidase. Int. J. Biol. Macromol. 2017, 103, 829–838. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Iacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Parenti, G.; Moracci, M.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase-A guide for the treatment of Pompe disease. Nat. Commun. 2017, 8, 1111. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, D.; De Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided. Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.L.; Sachett, L.G.; Pol-Fachin, L.; Verli, H. GROMOS96 43a1 performance in predicting oligosaccharide conformational ensembles within glycoproteins. Carbohydr. Res. 2010, 345, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Arthur, J. Olson Small Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Artemova, S.; Jaillet, L.; Redon, S. Automatic molecular structure perception for the universal force field. J. Comput. Chem. 2016, 37, 1191–1205. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Bhattacharjee, A.; Abul Farah, M.; Al-Hemaid, F.; Abou-Tarboush, F.M.; Al-Anazi, K.M.; Al-Anazi, F.S.M.; Lee, J. Molecular docking of the anticancer bioactive compound proceraside with macromolecules involved in the cell cycle and DNA replication. Genet. Mol. Res. 2016, 15, 15027829. [Google Scholar] [CrossRef]
- Qasim Khan, A.; Malik, A. A steroid from Calotropis procera. Phytochemistry 1989, 28, 2859–2861. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Heifets, A.; Lilien, R.H. LigAlign: Flexible ligand-based active site alignment and analysis. J. Mol. Graph. Model. 2010, 29, 93–101. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa-A GROMACS tool for MM-PBSA and its optimization for high-throughput binding energy calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Obiol-Pardo, C.; Rubio-Martinez, J. Comparative evaluation of MMPBSA and XSCORE to compute binding free energy in XIAP-peptide complexes. J. Chem. Inf. Model. 2007, 47, 134–142. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; Scientific Research; R Development Core Team: Vienna, Austria, 2008; Volume 2, ISBN 3-900051-07-0. [Google Scholar]
- Li, L.; Qu, C.; Wu, X.; Dai, J.; Lu, Y.; Gong, Y.; You, R.; Liu, Y. Patterns and levels of platelet glycosylation in patients with coronary heart disease and type 2 diabetes mellitus. J. Thromb. Thrombolysis 2018, 45, 56–65. [Google Scholar] [CrossRef]
- Jhong, C.H.; Riyaphan, J.; Lin, S.H.; Chia, Y.C.; Weng, C.F. Screening alpha-glucosidase and alpha-amylase inhibitors from natural compounds by molecular docking in silico. BioFactors 2015, 41, 242–251. [Google Scholar] [CrossRef]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef]
- Prottoy, N.I.; Ullah, A.; Sarkar, B.; Hossain, S.; Boby, A.S. Molecular Docking and Pharmacological Property Analysis of Antidiabetic Agents from Medicinal Plants of Bangladesh against Type II Diabetes: A Computational Approach. PharmaTutor 2019, 7, 6–15. [Google Scholar] [CrossRef]
- Abdallah, H.M.; Zakaria, E.M.; El-Halawany, A.M.; Mohamed, G.A.; Safo, M.K.; El-Bassossy, H.M. Psiadia punctulata major flavonoids alleviate exaggerated vasoconstriction produced by advanced glycation end products. PLoS ONE 2019, 14, e0222101. [Google Scholar] [CrossRef]
- Han, X.; Deng, Y.; Yu, J.; Sun, Y.; Ren, G.; Cai, J.; Zhu, J.; Jiang, G. Acarbose Accelerates Wound Healing via Akt/eNOS Signaling in db/db Mice. Oxid. Med. Cell. Longev. 2017, 2017, 7809581. [Google Scholar] [CrossRef] [Green Version]
- Kasturi, S.; Surarapu, S.; Uppalanchi, S.; Anireddy, J.S.; Dwivedi, S.; Anantaraju, H.S.; Perumal, Y.; Sigalapalli, D.K.; Babu, B.N.; Ethiraj, K.S. Synthesis and α-glucosidase inhibition activity of dihydroxy pyrrolidines. Bioorganic Med. Chem. Lett. 2017, 27, 2818–2823. [Google Scholar] [CrossRef]
- L’hadj, I.; Azzi, R.; Lahfa, F.; Koceir, E.A.; Omari, N. The nutraceutical potential of Lepidium sativum L. seed flavonoid-rich extract in managing metabolic syndrome components. J. Food Biochem. 2019, 43, 12725. [Google Scholar] [CrossRef]
- Ren, F.; Chen, L.; Xiong, S.; Tong, Q. Enhanced acarbose production by Streptomyces M37 using a two-stage fermentation strategy. PLoS ONE 2017, 12, e0166985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Cai, L.; Guo, Y.; Zhang, H.; Wang, P.; Yi, G.; Huang, Y. Calotropin activates YAP through downregulation of LATS1 in colorectal cancer cells. Onco. Targets. Ther. 2019, 12, 4047–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewi, R.T.; Maryani, F. Antioxidant and α-Glucosidase Inhibitory Compounds of Centella Asiatica. Procedia Chem. 2015, 17, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Kwon, C.S.; Son, K.H. Inhibition of alpha-glucosidase and amylase by luteolin, a flavonoid. Biosci. Biotechnol. Biochem. 2000, 64, 2458–2461. [Google Scholar] [CrossRef]
- Taha, M.; Shah, S.A.A.; Afifi, M.; Imran, S.; Sultan, S.; Rahim, F.; Khan, K.M. Synthesis, α-glucosidase inhibition and molecular docking study of coumarin based derivatives. Bioorg. Chem. 2018, 77, 586–592. [Google Scholar] [CrossRef]
- Kairys, V.; Baranauskiene, L.; Kazlauskiene, M.; Matulis, D.; Kazlauskas, E. Binding affinity in drug design: Experimental and computational techniques. Expert Opin. Drug Discov. 2019, 14, 755–768. [Google Scholar] [CrossRef]
- Kastritis, P.L.; Bonvin, A.M.J.J. On the binding affinity of macromolecular interactions: Daring to ask why proteins interact. J. R. Soc. 2012, 5, 20120835. [Google Scholar] [CrossRef]
- Tabussum, A.; Riaz, N.; Saleem, M.; Ashraf, M.; Ahmad, M.; Alam, U.; Jabeen, B.; Malik, A.; Jabbar, A. α-Glucosidase inhibitory constituents from Chrozophora plicata. Phytochem. Lett. 2013, 6, 614–619. [Google Scholar] [CrossRef]
- Yin, Z.; Zhang, W.; Feng, F.; Zhang, Y.; Kang, W. α-Glucosidase inhibitors isolated from medicinal plants. Food Sci. Hum. Wellness 2014, 3, 136–174. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, B.; Tan, C.; Huang, Q. α-Glucosidase inhibitors: Consistency of: In silico docking data with in vitro inhibitory data and inhibitory effect prediction of quercetin derivatives. Food Funct. 2019, 10, 6312–6321. [Google Scholar] [CrossRef]
- Taha, M.; Ismail, N.H.; Imran, S.; Wadood, A.; Rahim, F.; Saad, S.M.; Khan, K.M.; Nasir, A. Synthesis, molecular docking and α-glucosidase inhibition of 5-aryl-2-(6′-nitrobenzofuran-2′-yl)-1,3,4-oxadiazoles. Bioorg. Chem. 2016, 66, 117–123. [Google Scholar] [CrossRef]
- Hameed, S.; Kanwal; Seraj, F.; Rafique, R.; Chigurupati, S.; Wadood, A.; Rehman, A.U.; Venugopal, V.; Salar, U.; Taha, M.; et al. Synthesis of benzotriazoles derivatives and their dual potential as α-amylase and α-glucosidase inhibitors in vitro: Structure-activity relationship, molecular docking, and kinetic studies. Eur. J. Med. Chem. 2019, 183, 111677. [Google Scholar] [CrossRef]
- Etsassala, N.G.E.R.; Badmus, J.A.; Waryo, T.T.; Marnewick, J.L.; Cupido, C.N.; Hussein, A.A.; Iwuoha, E.I. Alpha-glucosidase and alpha-amylase inhibitory activities of novel abietane diterpenes from Salvia Africana-Lutea. Antioxidants 2019, 8, 421. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Zhang, Q.; Wang, W.; Huang, H.; Kang, W. α-glucosidase inhibitory active constituents contained in nutshell of Trapa acornis. Zhongguo Zhongyao Zazhi 2012, 37, 1408–1411. [Google Scholar] [CrossRef]
- Atta-ur-Rahman; Zareen, S.; Choudhary, M.I.; Akhtar, M.N.; Khan, S.N. α-glucosidase inhibitory activity of triterpenoids from Cichorium intybus. J. Nat. Prod. 2008, 71, 910–913. [Google Scholar] [CrossRef]
- Li, H.; Song, F.; Xing, J.; Tsao, R.; Liu, Z.; Liu, S. Screening and Structural Characterization of α-Glucosidase Inhibitors from Hawthorn Leaf Flavonoids Extract by Ultrafiltration LC-DAD-MSn and SORI-CID FTICR MS. J. Am. Soc. Mass Spectrom. 2009, 20, 1496–1503. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.S.; Escher, G.B.; Vieira do Carmo, M.; Azevedo, L.; Boscacci Marques, M.; Daguer, H.; Molognoni, L.; Inés Genovese, M.; Wen, M.; Zhang, L.; et al. A new analytical concept based on chemistry and toxicology for herbal extracts analysis: From phenolic composition to bioactivity. Food Res. Int. 2020, 132, 109090. [Google Scholar] [CrossRef]
- Banz, K.; Dinkel, R.; Hanefeld, M.; Schwanebeck, U. Evaluation of the potential clinical and economic effects of bodyweight stabilisation with acarbose in patients with type 2 diabetes mellitus: A decision-analytical approach. Pharmacoeconomics 1998, 13, 449–459. [Google Scholar] [CrossRef]
- Takahashi, K.; Tokuoka, M.; Kohno, H.; Sawamura, N.; Myoken, Y.; Mizuno, A. Comprehensive analysis of dipeptides in alcoholic beverages by tag-based separation and determination using liquid chromatography/electrospray ionization tandem mass spectrometry and quadrupole-time-of-flight mass spectrometry. J. Chromatogr. A 2012, 1242, 17–25. [Google Scholar] [CrossRef]
- Malik, A.; Jamil, U.; Butt, T.T.; Waquar, S.; Gan, S.H.; Shafique, H.; Jafar, T.H. In silico and in vitro studies of lupeol and iso-orientin as potential antidiabetic agents in a rat model. Drug Des. Devel. Ther. 2019, 13, 1501–1513. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wu, S.; Zhang, Q.; Yin, Z.; Zhang, L. α-Glucosidase inhibitory effect of anthocyanins from Cinnamomum camphora fruit: Inhibition kinetics and mechanistic insights through in vitro and in silico studies. Int. J. Biol. Macromol. 2019, 143, 696–703. [Google Scholar] [CrossRef]
- Fu, Y.; Zhao, J.; Chen, Z. Insights into the Molecular Mechanisms of Protein-Ligand Interactions by Molecular Docking and Molecular Dynamics Simulation: A Case of Oligopeptide Binding Protein. Comput. Math. Methods Med. 2018, 2018, 3502514. [Google Scholar] [CrossRef]
- Bhakat, S. Effect of T68A/N126Y mutations on the conformational and ligand binding landscape of Coxsackievirus B3 3C protease. Mol. Biosyst. 2015, 11, 2303–2311. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Munawar, S.; Windley, M.J.; Tse, E.G.; Todd, M.H.; Hill, A.P.; Vandenberg, J.I.; Jabeen, I. Experimentally validated pharmacoinformatics approach to predict hERG inhibition potential of new chemical entities. Front. Pharmacol. 2018, 9, 1035. [Google Scholar] [CrossRef]
- Varma, A.K.; Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of Drug-Designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Menéndez, C.A.; Accordino, S.R.; Gerbino, D.C.; Appignanesi, G.A. Hydrogen bond dynamic propensity studies for protein binding and drug design. PLoS ONE 2016, 11, e0165767. [Google Scholar] [CrossRef] [Green Version]
- Abuelizz, H.A.; Anouar, E.H.; Ahmad, R.; Nor Azman, N.I.I.; Marzouk, M.; Al-Salahi, R. Triazoloquinazolines as a new class of potent α-glucosidase inhibitors: In vitro evaluation and docking study. PLoS ONE 2019, 14, e0220379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, H.X.; Kusaka, A.; Kitakawa, C.K.; Onari, J.; Yamanaka, S.; Nakamura, H.; Takano, Y. Hydrogen bond donors and acceptors are generally depolarized in α-helices as revealed by a molecular tailoring approach. J. Comput. Chem. 2019, 40, 2043–2052. [Google Scholar] [CrossRef] [Green Version]
- Granchi, C.; Capecchi, A.; Del Frate, G.; Martinelli, A.; Macchia, M.; Minutolo, F.; Tuccinardi, T. Development and validation of a docking-based virtual screening platform for the identification of new lactate dehydrogenase inhibitors. Molecules 2015, 20, 8772–8790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alogheli, H.; Olanders, G.; Schaal, W.; Brandt, P.; Karlén, A. Docking of Macrocycles: Comparing Rigid and Flexible Docking in Glide. J. Chem. Inf. Model. 2017, 57, 190–202. [Google Scholar] [CrossRef]
- Ballante, F.; Marshall, G.R. An Automated Strategy for Binding-Pose Selection and Docking Assessment in Structure-Based Drug Design. J. Chem. Inf. Model. 2016, 56, 54–72. [Google Scholar] [CrossRef]
- Lagunin, A.; Filimonov, D.; Poroikov, V. Multi-Targeted Natural Products Evaluation Based on Biological Activity Prediction with PASS. Curr. Pharm. Des. 2010, 16, 1703–1717. [Google Scholar] [CrossRef] [Green Version]
- Filimonov, D.A.; Druzhilovskiy, D.S.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Dmitriev, A.V.; Pogodin, P.V.; Poroikov, V.V. Computer-aided prediction of biological activity spectra for chemical compounds: Opportunities and limitation. Biomed. Chem. Res. Methods 2018, 1, e00004. [Google Scholar] [CrossRef] [Green Version]
- Doan, T.N.; Thi, L.; Trungward, L.; Ducdistrict, T. Targeted proteins for diabetes drug design. Adv. Nat. Sci. Nanosci. Nanotechnol. 2012, 3, 013001. [Google Scholar] [CrossRef]
- Durrant, J.D.; Mccammon, J.A. Molecular dynamics simulations and drug discovery. J. Biol. 2011, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Behmard, E.; Abdolmaleki, P.; Taghdir, M. Understanding the inhibitory mechanism of BIT225 drug against p7 viroporin using computational study. Biophys. Chem. 2018, 233, 47–54. [Google Scholar] [CrossRef]
- Nichols, S.E.; Baron, R.; Ivetac, A.; McCammon, J.A. Predictive power of molecular dynamics receptor structures in virtual screening. J. Chem. Inf. Model. 2011, 51, 1439–1446. [Google Scholar] [CrossRef]
- Ul Haq, F.; Abro, A.; Raza, S.; Liedl, K.R.; Azam, S.S. Molecular dynamics simulation studies of novel β-lactamase inhibitor. J. Mol. Graph. Model. 2017, 74, 143–152. [Google Scholar] [CrossRef]
- Carugo, O. How root-mean-square distance (r.m.s.d.) values depend on the resolution of protein structures that are compared. J. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]
- Cholko, T.; Chen, W.; Tang, Z.; Chang, C. A Molecular Dynamics investigation of CDK8/CycC and Ligand binding: Conformational flexibilty and implication in drug discovery. J. Comput. Aided. Mol. Des. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinforma. Chem. 2015, 8, 37. [Google Scholar]
- Galzitskaya, O.V.; Garbuzynskiy, S.O. Entropy capacity determines protein folding. Proteins Struct. Funct. Genet. 2006, 63, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.W.; Liao, M.L.; Meng, X.L.; Somero, G.N. Structural flexibility and protein adaptation to temperature: Molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc. Natl. Acad. Sci. USA 2018, 115, 1274–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasner, E.; Hunter, C.A.; Kariko, K. Molecular Docking: A powerful approach for structure-based drug discovery. Natl. Inst. Health 2013, 70, 646–656. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Chaudhary, N.; Aparoy, P. Deciphering the mechanism behind the varied binding activities of COXIBs through Molecular Dynamic Simulations, MM-PBSA binding energy calculations and per-residue energy decomposition studies. J. Biomol. Struct. Dyn. 2017, 35, 868–882. [Google Scholar] [CrossRef]
- Gupta, A.; Chaudhary, N.; Aparoy, P. MM-PBSA and per-residue decomposition energy studies on 7-Phenyl-imidazoquinolin-4(5H)-one derivatives: Identification of crucial site points at microsomal prostaglandin E synthase-1 (mPGES-1) active site. Int. J. Biol. Macromol. 2018, 119, 352–359. [Google Scholar] [CrossRef]
- Hu, X.; Xie, J.; Hu, S.; Zhang, L.; Dong, Y. Exploration of the binding affinities between ecdysone agonists and EcR/USP by docking and MM-PB/GBSA approaches. J. Mol. Model. 2017, 23, 166. [Google Scholar] [CrossRef]
- Appiah-Kubi, P.; Soliman, M. Hybrid Receptor-Bound/MM-GBSA-Per-residue Energy-Based Pharmacophore Modelling: Enhanced Approach for Identification of Selective LTA4H Inhibitors as Potential Anti-inflammatory Drugs. Cell Biochem. Biophys. 2017, 75, 35–48. [Google Scholar] [CrossRef]
- Appiah-Kubi, P.; Soliman, M.E.S. Dual anti-inflammatory and selective inhibition mechanism of leukotriene A4 hydrolase/aminopeptidase: Insights from comparative molecular dynamics and binding free energy analyses. J. Biomol. Struct. Dyn. 2016, 34, 2418–2433. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Q.; Zhou, F.C.; Gao, F.; Bian, J.S.; Shan, F. Comparative evaluation of quercetin, isoquercetin and rutin as inhibitors of α-glucosidase. J. Agric. Food Chem. 2009, 57, 11463–11468. [Google Scholar] [CrossRef] [PubMed]
- Shanno, R.L. Rutin; A new drug for the treatment of increased capillary fragility. Am. J. Med. Sci. 1946, 211, 539–543. [Google Scholar] [CrossRef]
- Cherigo, L.; Martínez-Luis, S. Identification of Major α-Glucosidase Inhibitors from Stem Bark of Panamanian Mangrove Plant Pelliciera rhizophorae. Nat. Prod. Commun. 2019, 14, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Phan, H.V.T.; Duong, T.H.; Pham, D.D.; Pham, H.A.; Nguyen, V.K.; Nguyen, T.P.; Nguyen, H.H.; Nguyen, N.H.; Samang, P.; Phontree, K.; et al. Design and synthesis of new lupeol derivatives and their α-glucosidase inhibitory and cytotoxic activities. Nat. Prod. Res. 2020, 17, 1758095. [Google Scholar] [CrossRef]
- Lestari, W.; Dewi, R.T.; Kardono, L.B.S.; Yanuar, A. Docking sulochrin and its derivative as α-glucosidase inhibitors of Saccharomyces cerevisiae. Indones. J. Chem. 2017, 17, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Zahoor, M.; Bari, W.U.; Zeb, A.; Khan, I. Toxicological, anticholinesterase, antilipidemic, antidiabetic and antioxidant potentials of Grewia optiva Drummond ex Burret extracts. J. Basic Clin. Physiol. Pharmacol. 2019, 31, 20190220. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound Names | Sources | PubChem ID | Mol. Weight | Refs. |
|---|---|---|---|---|---|
| 1 | Isorhamnetin-3-O-rutinoside | Stem/Latex | 5481663 | 624.54 | [19] |
| 2 | Isorhamnetin-3-O-robinobioside | Stem/Latex | 5491808 | 624.5 | [19,20] |
| 3 | Calotropagenin | Leaf/Latex | 212348 | 404.5 | [18] |
| 4 | Calotoxin | Latex | 56840852 | 404.5 | [19,22] |
| 5 | Uscharin | Latex/Leaf | 11261800 | 587.72 | [19,25] |
| 6 | Voruscharin | Latex | 44387915 | 589.74 | [19] |
| 7 | 2,7,10-trimethyldodecane | Stem Bark | 93447 | 212.41 | [19] |
| 8 | Luteolin | Leaf | 15661823 | 300.26 | [19] |
| 9 | Ursolic Acid | Leaf | 64945 | 456.7 | [19] |
| 10 | β-amyrin | Latex/Root | 73145 | 426.72 | [19,45] |
| 11 | Syriogenin | Leaf | 11870470 | 390.51 | [18,19] |
| 12 | Lactucerol | Latex | 115250 | 426.7 | [19] |
| 13 | Octadecenamide | Stem Bark | 6443016 | 281.5 | [19] |
| 14 | Z-13 docosinamide | Stem/Latex | 5365371 | 337.6 | [19] |
| 15 | Tyranton | Leaf | 31256 | 116.16 | [19] |
| 16 | 1-heptadecene | Leaf | 23217 | 238.5 | [19] |
| 17 | Taraxasterol | Root | 344468 | 468.8 | [19,45] |
| 18 | Benzoyllineolone | Root bark | 5322013 | 468.6 | [19,45] |
| 19 | 3-epimoretenol | Latex | 604951 | 426.72 | [19] |
| 20 | 1-pentadecene | Leaf | 25913 | 210.4 | [19] |
| 21 | Isobutylnonane | Stem/Latex | 545936 | 184.36 | [19] |
| 22 | α-amyrin | Root bark | 73170 | 426.72 | [19,45] |
| 23 | Glibenclamide | Root | 3488 | 494 | [24] |
| 24 | Apigenin-7-0-glucoside | Leaf/Root | 5280704 | 432.38 | [18,19] |
| 25 | Thioacetic acid | Leaf | 10484 | 76.12 | [18,19] |
| 26 | kaempferol-7-0-glucoside | Leaf | 10095180 | 448.38 | [19,25] |
| 27 | Quercetin-3-rutinoside | Latex | 5280805 | 610.5 | [19] |
| 28 | Calotropin | Leaf/Stem/latex | 16142 | 532.6 | [19] |
| 29 | Beta sitosterol | Stem Bark | 222284 | 414.71 | [19,25] |
| 30 | Benzoylisolineolone | Root bark | 9982084 | 468.58 | [19] |
| 31 | Calactin | Leaf | - | 523.6 | [19] |
| 32 | Procesterol | Undried flower | - | 428.69 | [46] |
| Extracted Compounds | Binding Energies (kJ/mol) | Hydrogen Bonding Interacting Residues and Bond Lengths (Å) | Hydrophobic Bond Interacting Residues |
|---|---|---|---|
| Taraxasterol | −40.2 | Leu677 (3.07), Leu678 (3.32) | Asp282, Leu283, Ala284, Trp376, Trp481, Met519, Ser523, Phe525, Asp616, Phe649, Leu650, Ser676 |
| Voruscharin | −39.3 | Arg281 (3.07), Asp616 (2.36), Leu678 (3.23) | Asp282, Trp376, Trp481, Met519, Ala655, Phe649, Leu650, Ser676, Leu677 |
| Alpha-amyrin | −37.7 | Phe525 (3.17) | Asp282, Trp376, Trp481, Ser523, Asp524, Ala555, Asp616, Leu650, Phe649 |
| 3-epimoretenol | −36.8 | None | Asp282, Trp376, Trp481, Met519, Asn524, Phe525, Phe649 |
| Lactucerol | −36.4 | None | Asp282, Trp376, Trp481, Asn524, Phe525, Ala555, Phe649, Leu650, Ser676 |
| Beta-sitosterol | −36.4 | Asn524 (3.11) | Asp282, Trp376, Leu404, Trp481, Ser523, Asn524, Phe525, Ala555, Asp616, Phe649, Leu650, Ser676 |
| Beta-amyrin | −36.0 | None | Asp282, Trp376, Trp481, Asn524, Phe525, Ala555, Phe649, Leu650, Asp616, Ser676 |
| Apigenin-7-0-glucoside | −36.0 | Asp404 (2.61, 2.94), Asn524 (2.92), Arg600 (2.94, 3.17), Asp616 (2.87, 3.30), His674 (3.22) | Asp282, Trp376, Leu405, Trp481, Ile441, Asp518, Met519, Phe525, Ala555, Phe649 |
| Uscharin | −35.1 | Asp616 (2.76) | Asp282, Trp376, Trp481, Asn524, Phe525, Phe649, Leu650, Asp616, Ser676, Leu677, Leu678 |
| Syriogenin | −35.1 | Arg281 (3.16), Asp282 (3.10), Asp616 (2.88), Leu677 (3.27) | Trp376, Trp481, Met519, Asn524, Phe525, Ala555, Phe649, Leu650, Phe649, Leu650, Ser676 |
| Quercetin-3-rutinoside | −34.7 | Asp282 (2.74,3.15,3.16), Asp404 (2.44), Asp518 (2.94), Ser523 (3.08), Arg600 (2.67, 3.15), Asp616 (3.04, 3.19), His674 (2.91) | Leu283, Ala284, Trp376, Trp481, Trp516, Met519, Asn524, Phe525, Phe649, Leu650 |
| Glibenclamide | −34.7 | Arg281 (3.02), Asp616 (2.85, 3.01) | Asp282, Leu283, Trp376, Asp404, Ile441, Trp481, Asn524, Phe525, Asp518, Ala555, Phe649, His674 |
| Benzoyllineolone | −34.7 | Asp282 (2.81) | Leu283, Ala284, Trp376, Trp481, Phe525, Ala555, Asp616, Phe649, Leu650 |
| Kaempferol-7-0-glucoside | −34.3 | Arg281 (3.20), Asp282 (2.91), Asp404 (3.02), Ser523 (3.07, 2.74), Asn524 (2.70, 3.00) | Leu283, Trp376, Ile441, Trp481, Phe525, Asp518, Trp516, Met519, Ala555, Asp616, Phe649 |
| Ursolic acid | −34.3 | None | Asp282, Trp376, Trp481, Asn518, Phe525, Ala555, Arg600, Asp616, Phe649, Ser676 |
| Isorhamnetin-3-O-rutinoside | −34.3 | Asp282 (2.82), Asp404 (3.03), Trp481 (3.32), Asp518 (2.81, 3.07), Arg600 (3.25), Asp616 (2.99) | Asp282, Leu283, Trp376, Ile441, Trp481, Asn524, Phe525, Asp518, Ala555, Phe649, His674 |
| Isorhamnetin-3-O-robinobioside | −34.3 | Asp282 (2.82), Asp404 (3.03), Trp481 (3.32), Asp518 (2.81, 3.07), Arg600 (3.25), Asp616 (2.99) | Leu283, Trp376, Leu405, Ile441, Phe525, Trp613, Leu650, Ser676 |
| Calotoxin | −34.3 | Asp282 (3.15), Asn524 (2.86), Phe525 (2.79), Asp616 (2.71) | Arg281, Leu283, Ala284, Trp376, Ala555, Leu650 |
| Acarbose | −34.3 | Asp282 (2.78,2.82,2.99), Asp404 (2.70, 2.86), Asn524 (2.80), Phe525 (2.92), Arg600 (2.81, 2.83), Asp616 (2.70, 2.80), His674 (3.05) | Asp281, Leu283, Ala284, Trp376, Leu405, Ile441, Trp481, Trp516, Asp518, Met519, Ala555, Trp613, Phe649 |
| Calactin | −33.5 | Trp618 (3.17) | Arg281, Asp282, Ala284, Asn524, Phe525, Arg527, Ala555, Asp616, Leu650 |
| Calotropin | −33.5 | Trp618 (3.21) | Arg281, Asp282, Ala284, Asn524, Phe525, Arg527, Ala555, Asp616, Leu650 |
| Procesterol | −33.1 | Asp282 (2.91,3.11), Arg600 (2.99), Asp616 (3.14) | Trp376, Met519, Phe525, Trp618, Phe649, Leu650, Gly651, Ser676, Leu677, Leu678 |
| Benzoylisolineolone | −33.1 | Arg281 (3.16), Ala284 (2.99) | Asp282, Leu283, Ala284, Trp376, Phe525, Phe649, Leu650 |
| Calotropagenin | −32.6 | Asp91 (3.14), Asp95 (3.26) | Ala93, Lys96, Ala97, Ile98, Tyr110, Pro125, Trp126, Arg275 |
| luteolin | −31.4 | Asp282 (3.15), Asp404 (2.86), Ser523 (3.13), His674 (2.96) | Trp376, Trp481, Trp516, Asp518, Met519, Phe525, Asp616, Phe649 |
| 2,7,10-trimethyldodecane | −23.4 | None | Trp376, Leu405, Trp481, Ile441, Asp518, Met519, Phe525, Ala555, Asp616, Phe649, Leu677 |
| Octadecenamide | −21.3 | Asp518 (3.23), Asp616 (3.26) His674 (3.16) | Trp376, Phe525, Trp613, Phe649, Leu650, Ser676 Leu677, Leu678 |
| 1-_pentadecene | −21.3 | None | Trp376, Leu405, Trp481, Ile441, Asp518, Met519, Phe525, Ala555, Asp616, Phe649, Leu677 |
| Z-13_docosinamide | −20.9 | None | Asp282, Trp376, Leu405, Trp481, Ile441, Asp518, Met519, Phe525, Asp616, Phe649, Leu677 |
| Isobutylnonane | −20.5 | None | Asp282, Trp376, Asp404, Trp481, Asp518, Met519, Phe525, Arg600, Asp616, Phe649, Leu677 |
| 1-heptadecene | −19.7 | None | Trp376, Asp404, Trp481, Asp518, Met519, Phe525, Arg600, Asp616, Phe649, Leu677 |
| Tyranton | −19.2 | Trp481 (3.21), Asp518 (2.92) Arg600 (3.03) | Trp376, Asp404, Leu405, Trp481, Trp516, Met519, Asp616, Phe649, His674 |
| Thioacetic acid | −10.9 | His674 (3.01) | Trp516, Asp518, Trp613, Asp616, Phe649 |
| Compound | Pa | Pi | Activity |
|---|---|---|---|
| Taraxasterol | 0.200 | 0.005 | α-Glucosidase inhibitor |
| 0.141 | 0.069 | Antidiabetic type 1 | |
| 0.367 | 0.008 | Hydroxysteroid dehydrogenase inhibitor | |
| 0.332 | 0.009 | Protein tyrosine phosphate inhibitor | |
| 0.226 | 0.005 | 17-Beta-hydroxysterol dehydrogenase inhibitor | |
| 3-epimoretenol | 0.142 | 0.012 | Alpha glucosidase activity |
| 0.057 | 0.029 | 17-Beta-hydroxysterol dehydrogenase inhibitor | |
| 0.128 | 0.113 | Antidiabetic type 2 | |
| Lactucerol | 0.200 | 0.050 | α-Glucosidase inhibitor |
| Syriogenin | 0.102 | 0.029 | α-Glucosidase inhibitor |
| Isorhamnetin-3-O-robinobioside | 0.818 | 0.001 | α-Glucosidase inhibitor |
| Calotoxin | 0.101 | 0.029 | α-Glucosidase inhibitor |
| Compound | Van der Waals Energy | Electrostatic Energy | Polar Solvation Energy | SASA Energy | Binding Energy |
|---|---|---|---|---|---|
| Taraxasterol | −102.625 ± 17.227 | −2.795 ± 6.568 | 35.103 ± 11.322 | −9.808 ± 1.483 | −80.125 ± 15.326 |
| Syriogenin | −102.534 ± 13.538 | −27.083 ± 21.100 | 56.247 ± 29.946 | −9.769 ± 1.843 | −83.139 ± 16.039 |
| Isorhamnetin-3-O-robinobioside | −203.397 ± 18.850 | −141.376 ± 24.067 | 252.953 ± 36.473 | −20.17 ± 1.577 | −111.99 ± 30.828 |
| Calotoxin | −114.182 ± 24.776 | −14.063 ± 18.510 | 55.190 ± 46.644 | −10.91 ± 2.923 | −83.963 ± 47.232 |
| Acarbose | −155.148 ± 26.589 | 413.658 ± 50.519 | 272.582 ± 49.072 | −17.75 ± 1.949 | 513.34 ± 35.886 |
| Name of Compound | IUPAC Name | 2D Structure |
| Taraxasterol | (3S,4aR,6aR,6aR,6bR,8aR,12S,12aR,14aR,14bR)-4,4,6a,6b,8a,12,14b-heptamethyl-11-methylidene-1,2,3,4a,5,6,6a,7,8,9,10,12,12a,13,14,14a-hexadecahydropicen-3-ol |  |
| Syriogenin | 3-[(3S,5S,8R,9S,10S,12R,13S,14S,17R)-3,12,14-trihydroxy-10,13-dimethyl-1,2,3,4,5,6,7,8,9,11,12,15,16,17-tetradecahydrocyclopenta[a]phenanthren-17-yl]-2H-furan-5-one |  |
| Isorhamnetin-3-O-robinobioside | 5,7-dihydroxy-2-(4-hydroxy-3-methoxyphenyl)-3-[(3R,4S,5R,6R)-3,4,5-trihydroxy-6-[[(2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one |  |
| Calotoxin | (1S,3R,5S,7R,8S,9R,10S,12R,14R,18R,19R,22S,23R)-8,9,10,22-tetrahydroxy-7,18-dimethyl-19-(5-oxo-2H-furan-3-yl)-4,6,11-trioxahexacyclo [12.11.0.03,12.05,10.015,23.018,22]pentacosane-14-carbaldehyde |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adinortey, C.A.; Kwarko, G.B.; Koranteng, R.; Boison, D.; Obuaba, I.; Wilson, M.D.; Kwofie, S.K. Molecular Structure-Based Screening of the Constituents of Calotropis procera Identifies Potential Inhibitors of Diabetes Mellitus Target Alpha Glucosidase. Curr. Issues Mol. Biol. 2022, 44, 963-987. https://doi.org/10.3390/cimb44020064
Adinortey CA, Kwarko GB, Koranteng R, Boison D, Obuaba I, Wilson MD, Kwofie SK. Molecular Structure-Based Screening of the Constituents of Calotropis procera Identifies Potential Inhibitors of Diabetes Mellitus Target Alpha Glucosidase. Current Issues in Molecular Biology. 2022; 44(2):963-987. https://doi.org/10.3390/cimb44020064
Chicago/Turabian StyleAdinortey, Cynthia A., Gabriel B. Kwarko, Russell Koranteng, Daniel Boison, Issaka Obuaba, Michael D. Wilson, and Samuel K. Kwofie. 2022. "Molecular Structure-Based Screening of the Constituents of Calotropis procera Identifies Potential Inhibitors of Diabetes Mellitus Target Alpha Glucosidase" Current Issues in Molecular Biology 44, no. 2: 963-987. https://doi.org/10.3390/cimb44020064
APA StyleAdinortey, C. A., Kwarko, G. B., Koranteng, R., Boison, D., Obuaba, I., Wilson, M. D., & Kwofie, S. K. (2022). Molecular Structure-Based Screening of the Constituents of Calotropis procera Identifies Potential Inhibitors of Diabetes Mellitus Target Alpha Glucosidase. Current Issues in Molecular Biology, 44(2), 963-987. https://doi.org/10.3390/cimb44020064