
An Insight into the Structural Requirements and Pharmacophore Identification of Carbonic Anhydrase Inhibitors to Combat Oxidative Stress at High Altitudes: An In-Silico Approach

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
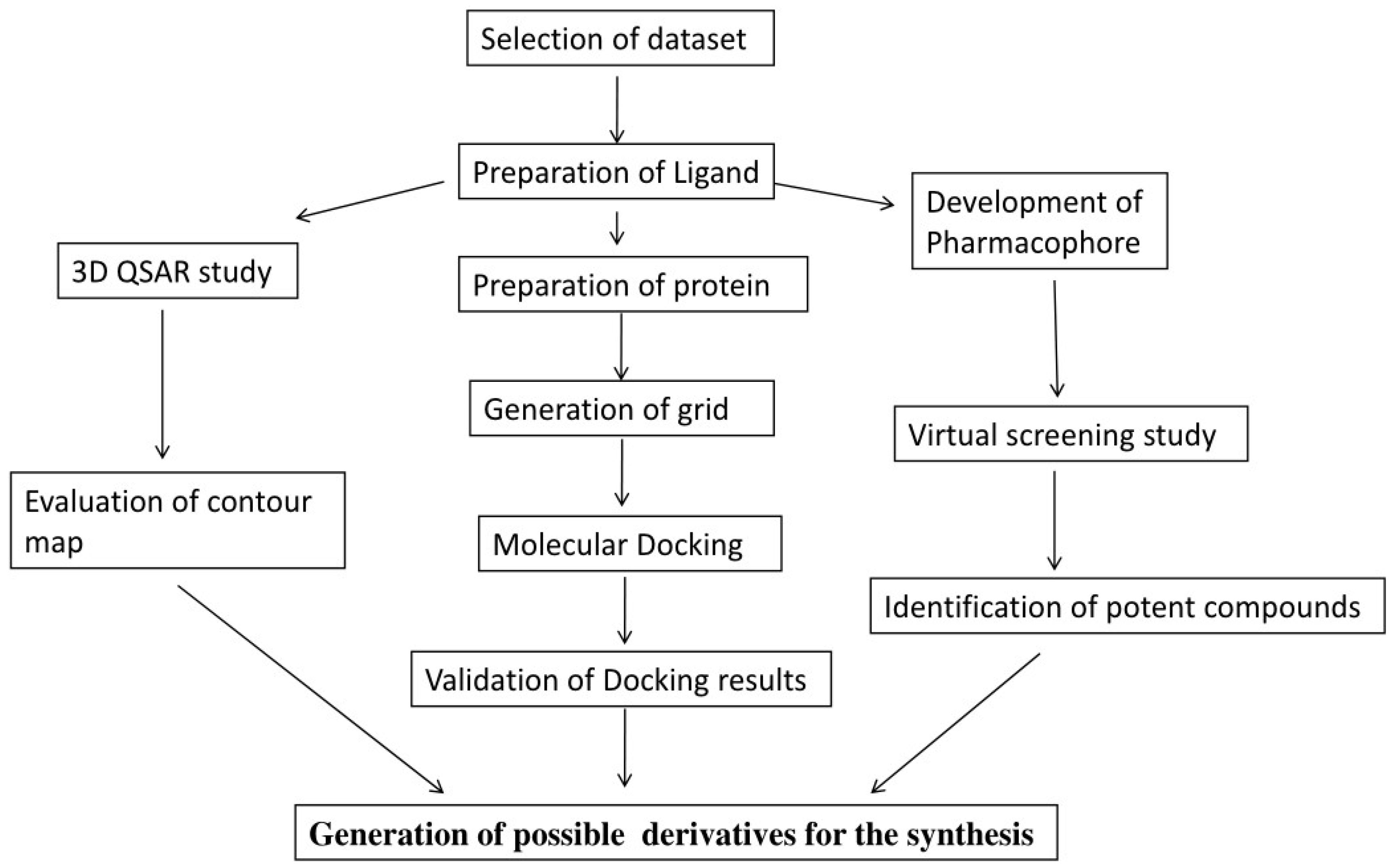
2. Materials and Methods
2.1. Collection of Data Set
2.2. Preparation of Ligands
2.3. Pharmacophore Mapping
2.4. Pharmacophore Hypothesis Generation
2.5. An Atom Based 3D-QSAR
2.6. Virtual Screening
2.7. Molecular Docking
2.8. ADME Properties Prediction
3. Results and Discussion
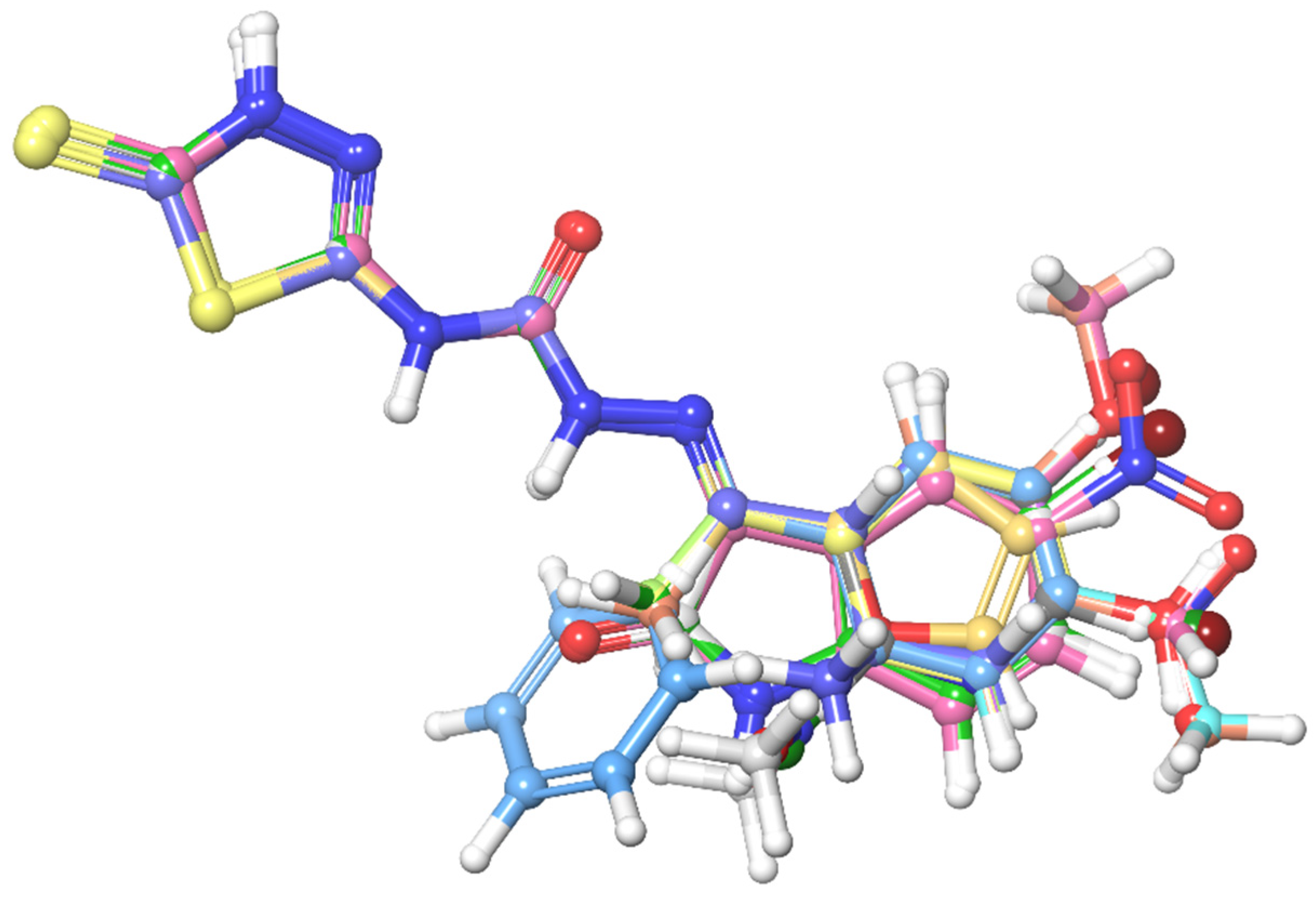
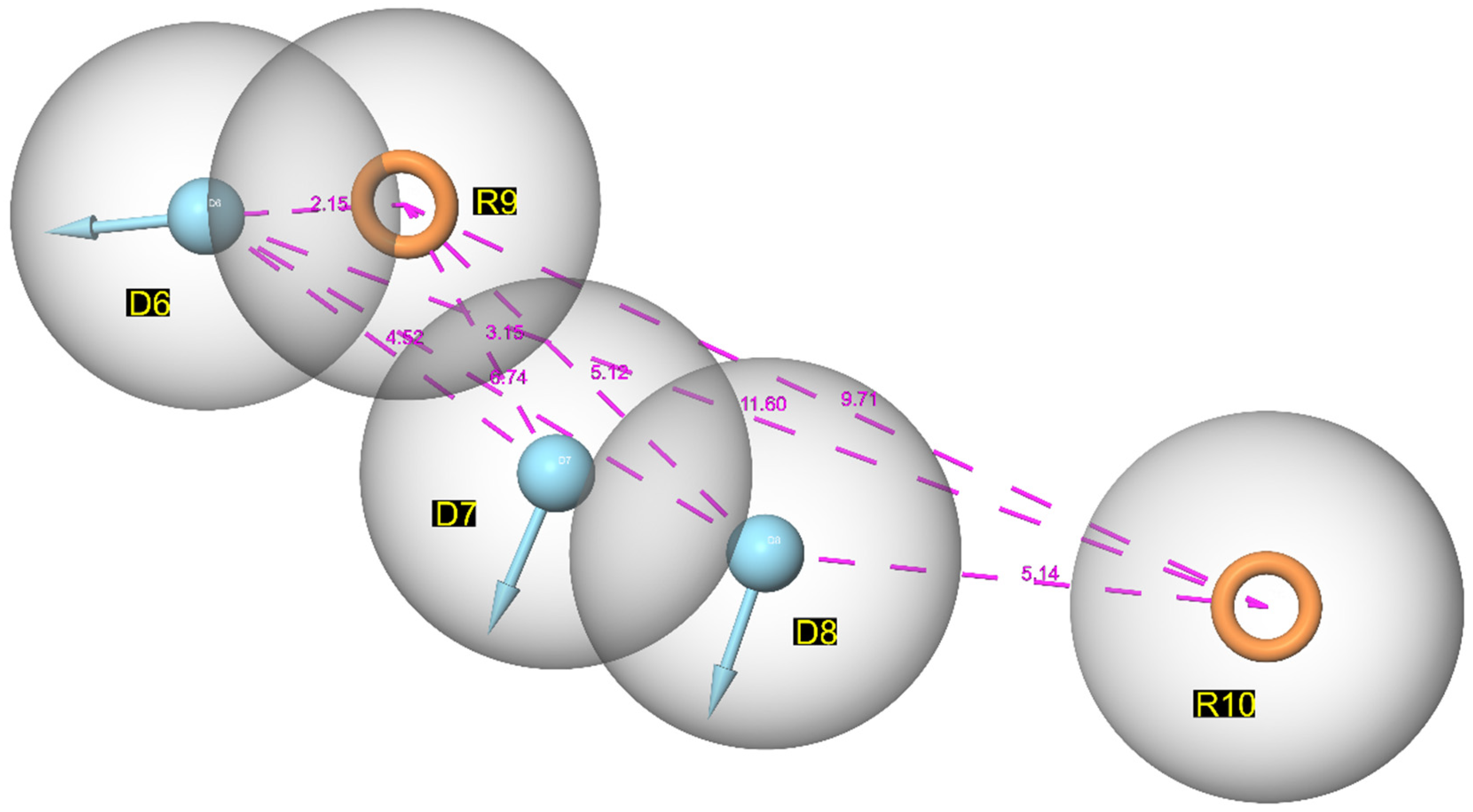
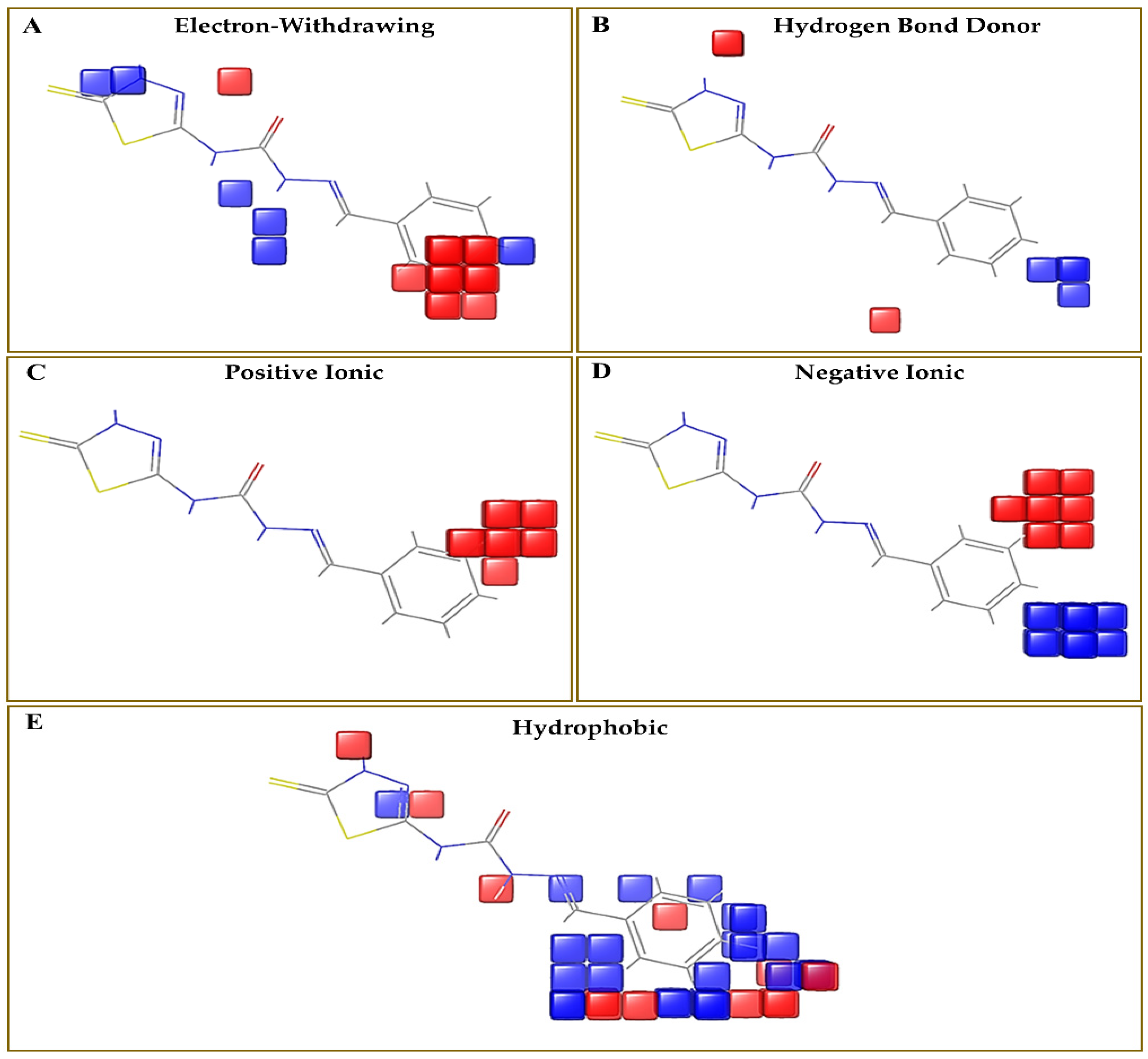
3.1. Pharmacophore Mapping: Selection of the Best Pharmacophore Hypothesis
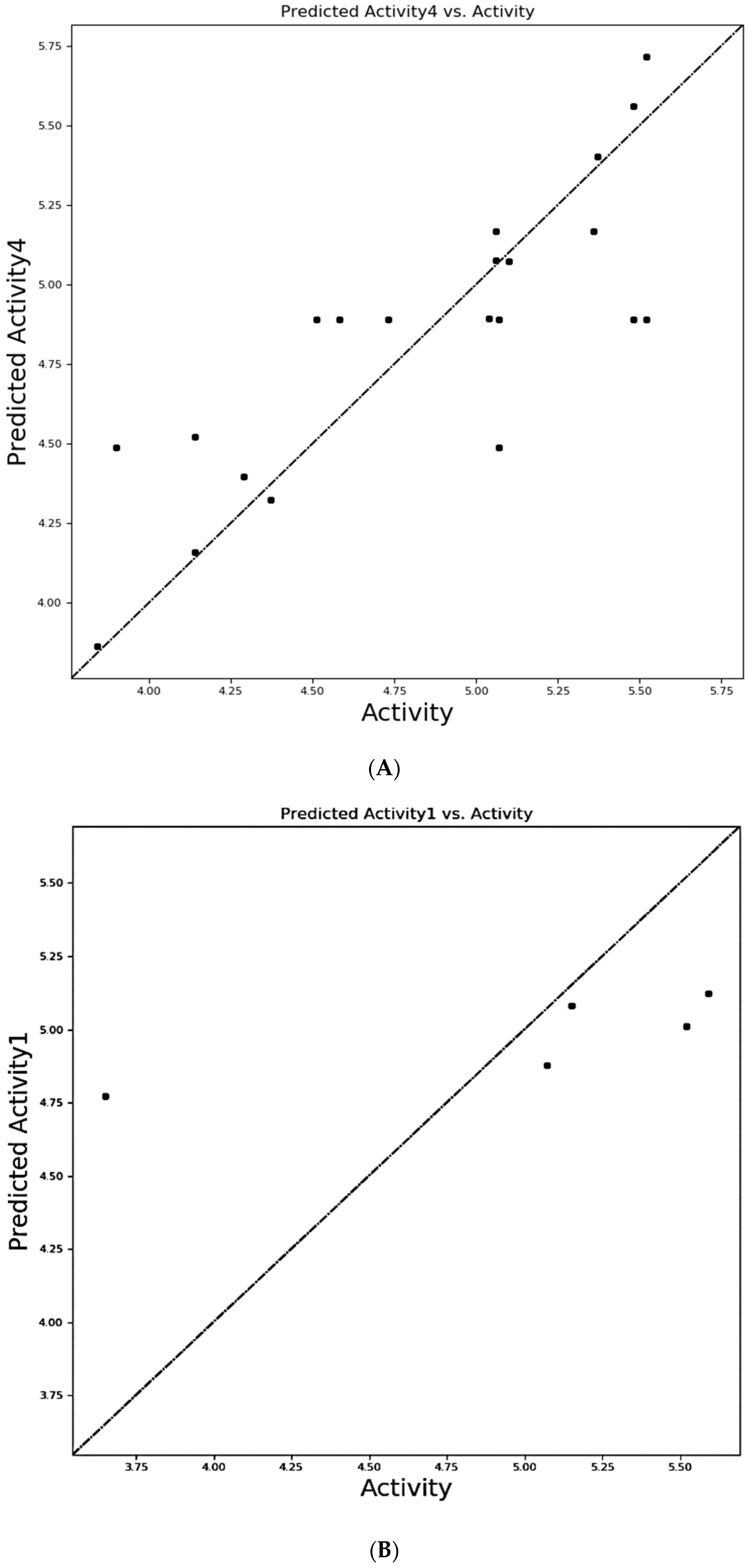
3.2. Selection of Atom Based QSAR Model
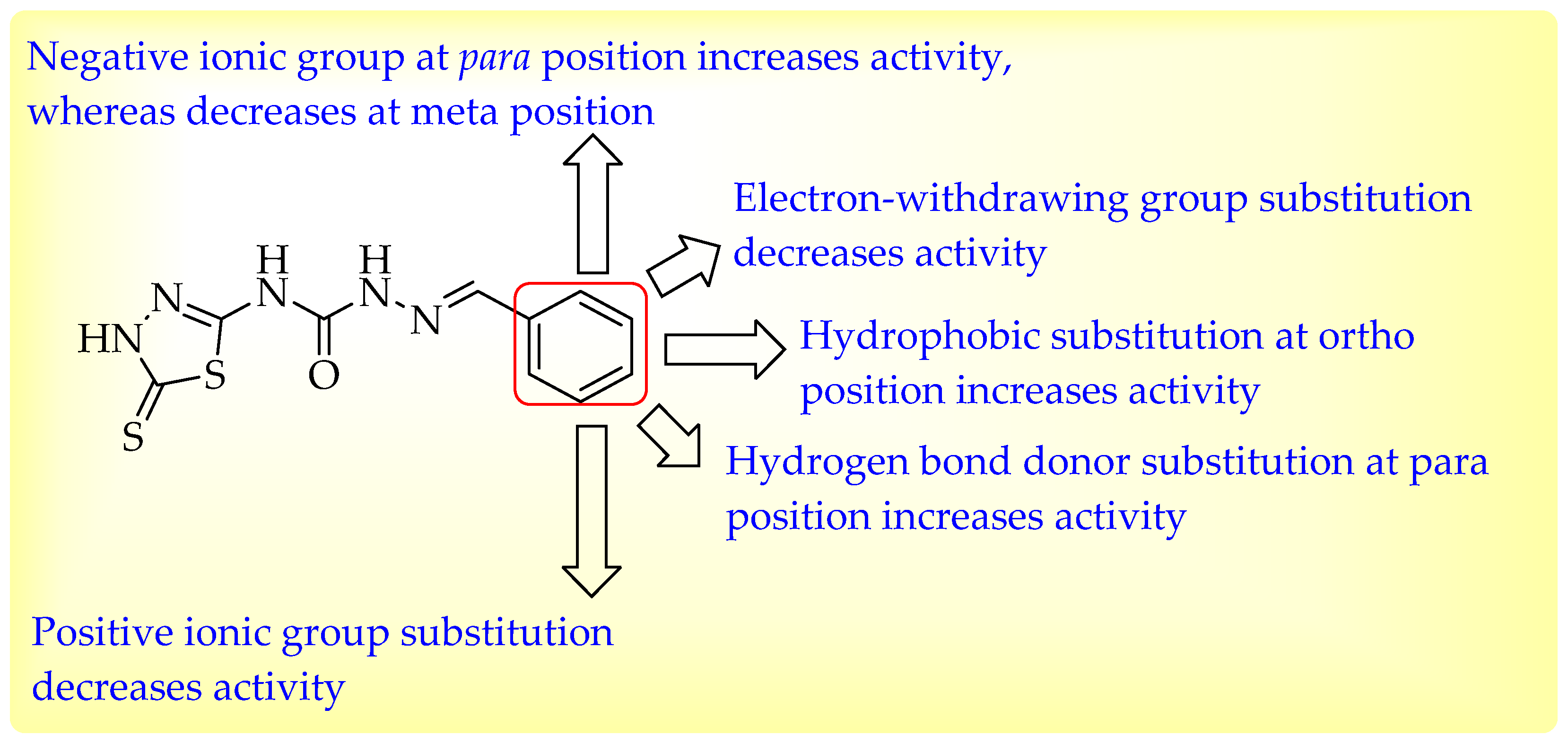
3.3. Evaluation of Contour Map
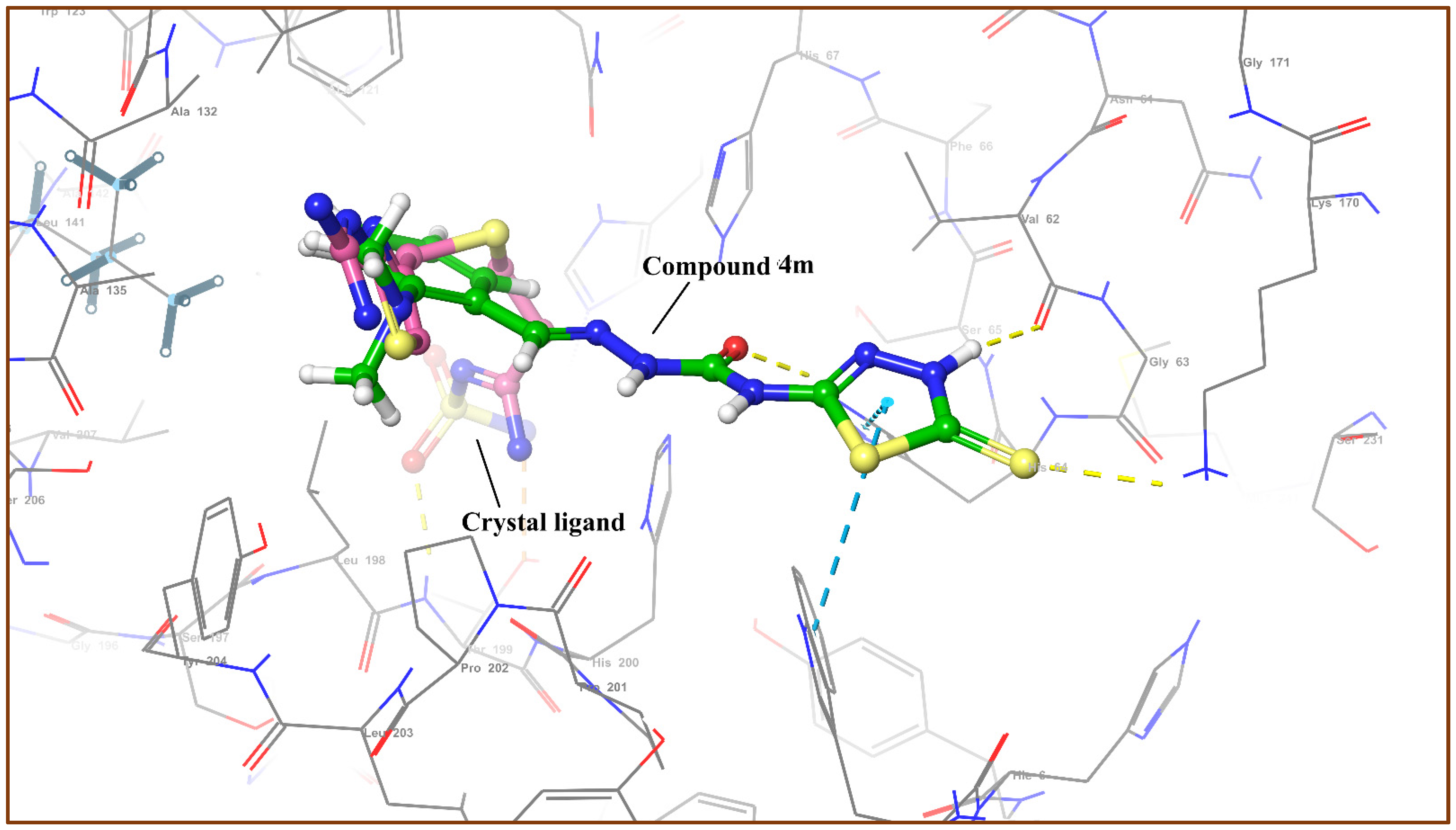
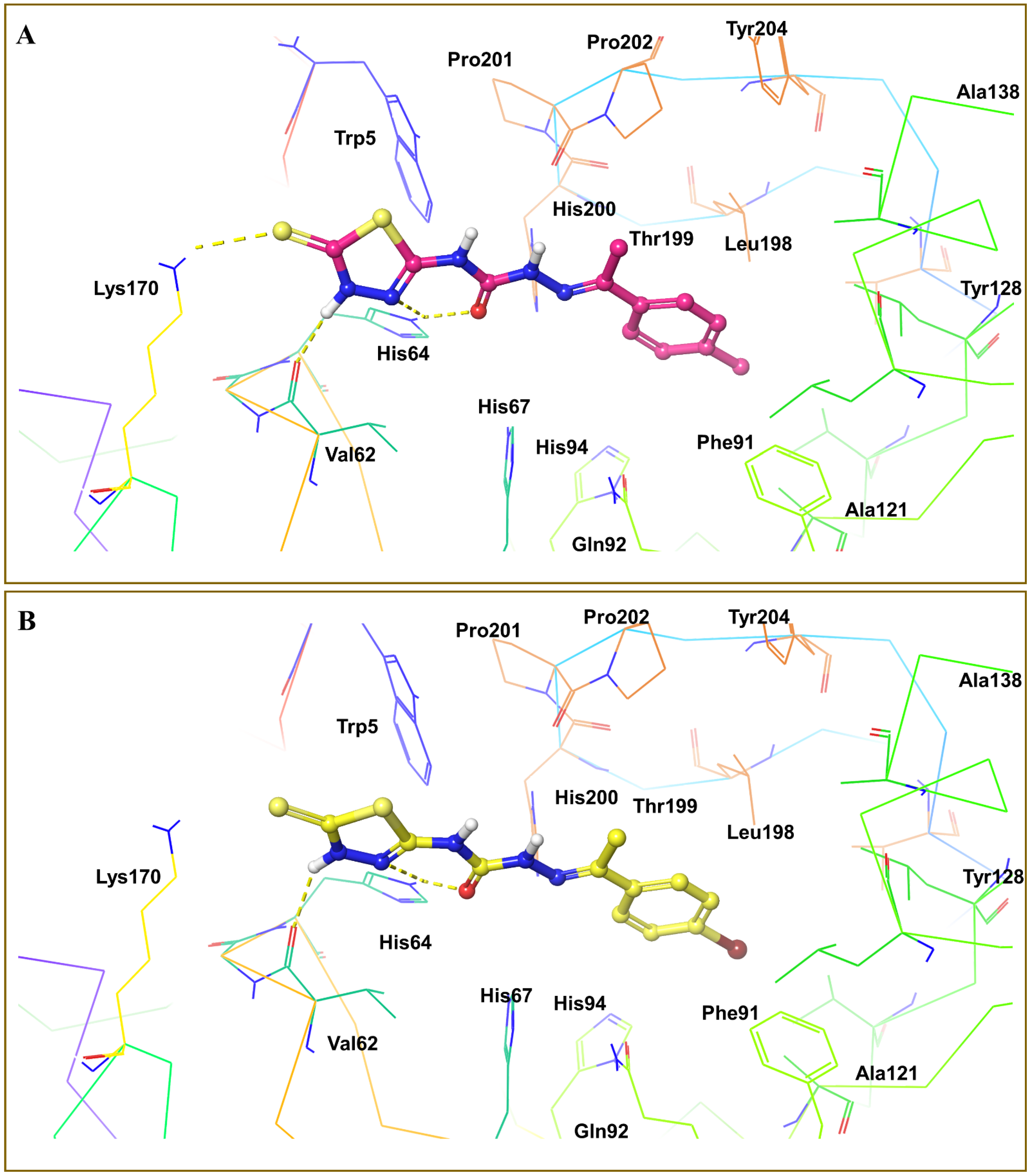
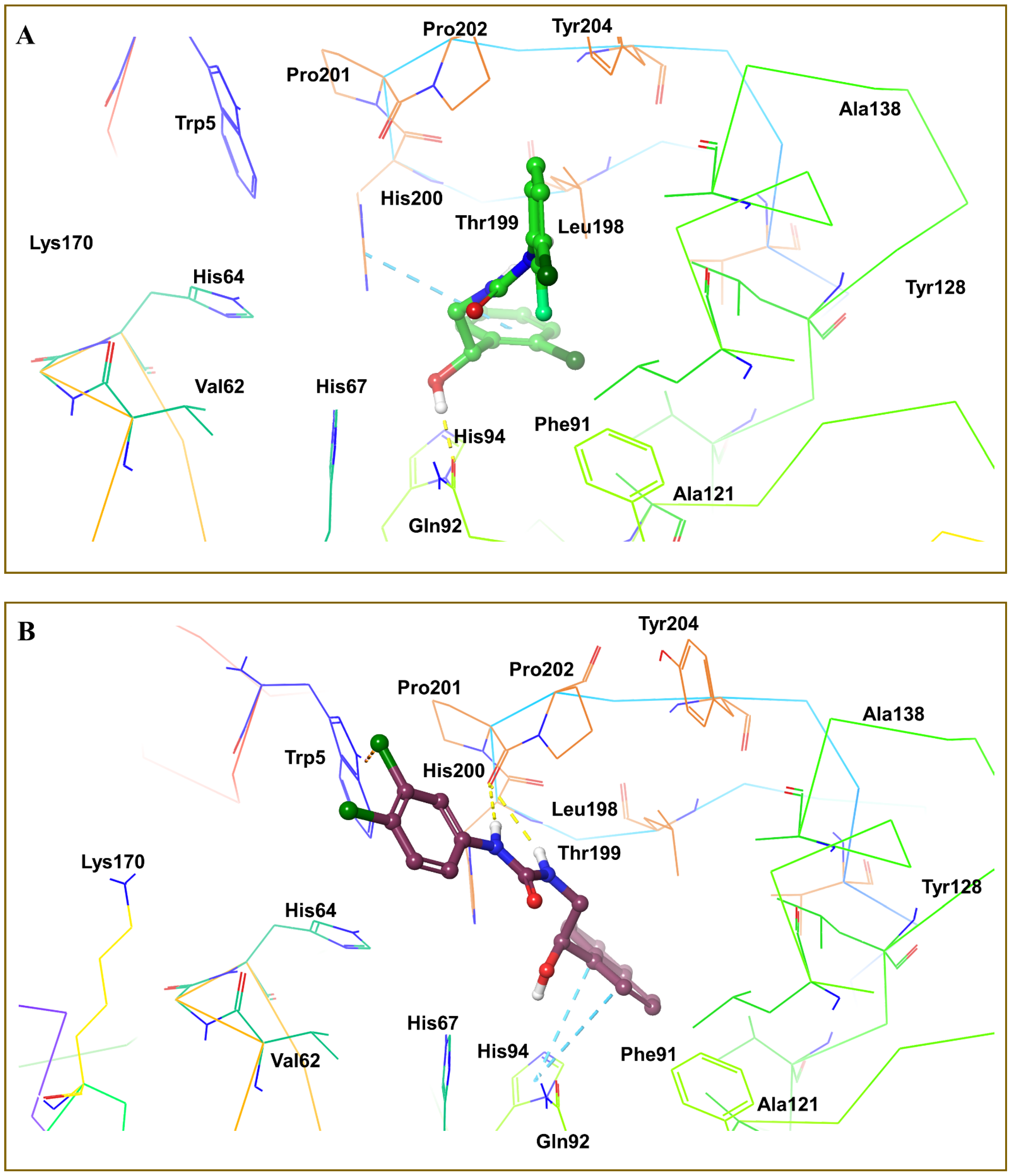
3.4. Molecular Docking Analysis
3.5. Virtual Screening
3.6. ADME Properties Prediction
3.7. MMGBSA-Based Rescoring
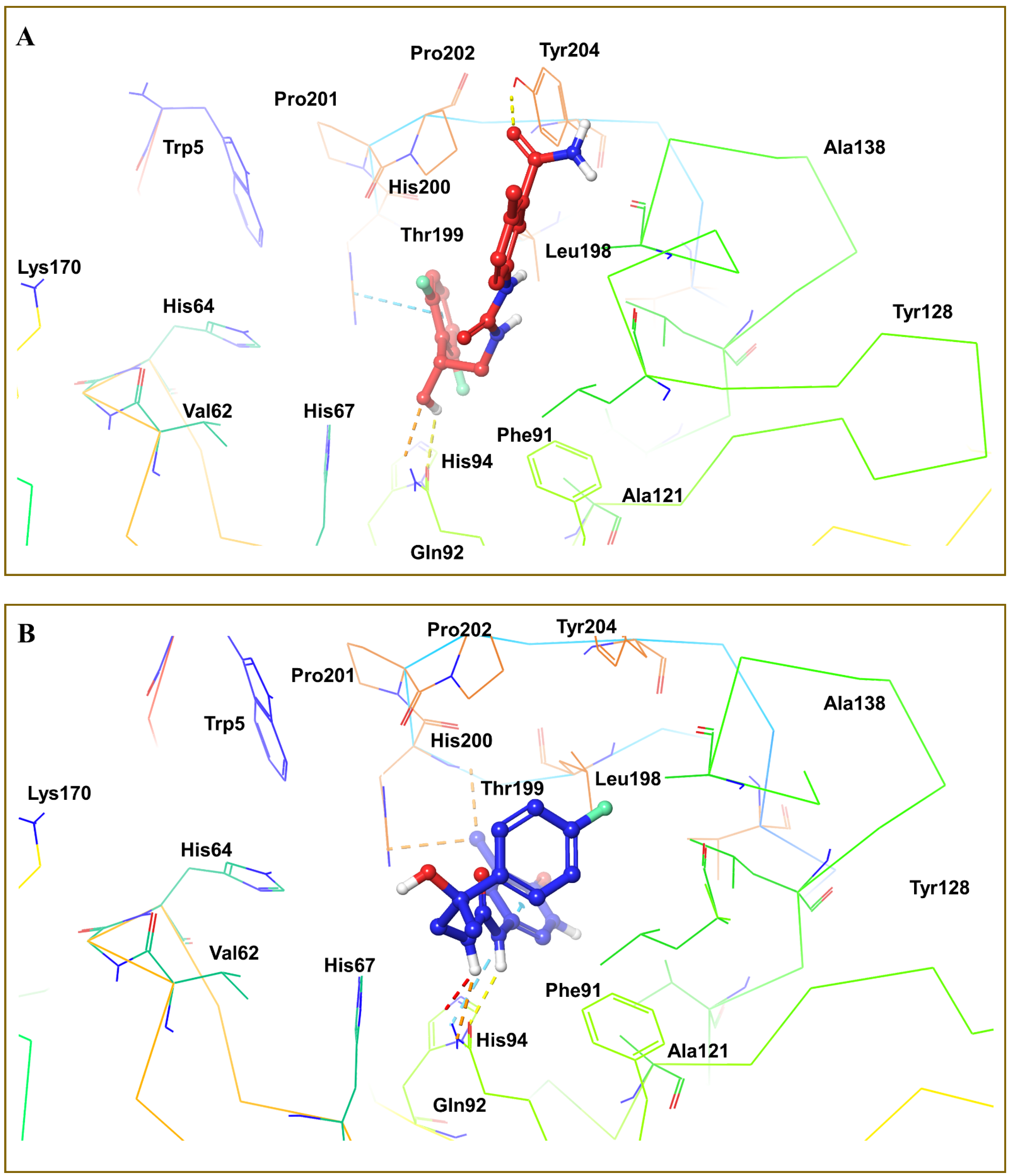
4. Optimization of Novel Ligands
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [Green Version]
- Therond, P. Dommagescréés aux biomolécules (lipides, protéines, ADN) par le stress oxydant [Oxidative stress and damages to biomolecules (lipids, proteins, DNA)]. Ann. Pharm. Fr. 2006, 64, 383–389. [Google Scholar] [CrossRef]
- Pacifici, R.E.; Davies, K.J. Protein, lipid and DNA repair systems in oxidative stress: The free-radical theory of aging revisited. Gerontology 1991, 37, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Aslam, S.; Gupta, V. Carbonic Anhydrase Inhibitors. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557736/ (accessed on 10 January 2022).
- Supuran, C.T. An update on drug interaction considerations in the therapeutic use of carbonic anhydrase inhibitors. Exp. Opin. Drug Metabol. Tox. 2020, 16, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Supuran, C.T. Treatment of sleep apnea with a combination of a carbonic anhydrase inhibitor and an aldosterone antagonist: A patent evaluation of CA2958110 and IN6616DEN2012. Exp. Opin. Ther. Pat. 2018, 28, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Pandey, R.K.; Kumbhar, B.V.; Sundar, S.; Kunwar, A.; Prajapati, V.K. Structure-based virtual screening, molecular docking, ADMET and molecular simulations to develop benzoxaborole analogs as potential inhibitor against Leishmania donovani trypanothione reductase. J. Recept. Signal Transduct. 2017, 37, 60–70. [Google Scholar] [CrossRef]
- Bakonyi, T.; Radak, Z. High Altitude and Free Radicals. J. Sports Sci. Med. 2004, 3, 64–69. [Google Scholar]
- Supuran, C.T. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert. Opin. Emerg. Drugs 2012, 17, 11–15. [Google Scholar] [CrossRef]
- Forwand, S.A.; Landowne, M.; Follansbee, J.N.; Hansen, J.E. Effect of acetazolamide on acute mountain sickness. N. Engl. J. Med. 1968, 279, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Secil, D.; Tugba, K.U.; Clemente, C.; Claudiu, T.S.; Ozen, O.G. Is carbonic anhydrase inhibition useful as a complementary therapy of Covid-19 infection? , J. Enzyme Inhib. Med. Chem. 2021, 36, 1230–1235. [Google Scholar] [CrossRef]
- Sechi, M.; Innocenti, A.; Pala, N.; Rogolina, D.; Carcelli, M.; Scozzafava, A.; Supuran, C.T. Inhibition of α-class cytosolic human carbonic anhydrases I, II, IX and XII, and β-class fungal enzymes by carboxylic acids and their derivatives: New isoform-I selective nanomolar inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5801–5806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorekova, S.; Zatovicova, M.; Pastorek, J. Cancer-associated carbonic anhydrases and their inhibition. Curr. Pharm. Des. 2008, 14, 685–698. [Google Scholar] [CrossRef]
- Carradori, S.; Mollica, A.; De Monte, C.; Ganese, A.; Supuran, C.T. Nitric oxide donors and selective carbonic anhydrase inhibitors: A dual pharmacological approach for the treatment of glaucoma, cancer and osteoporosis. Molecules 2015, 20, 5667–5679. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.K.; Sharma, S.; Vaidya, A.; Ravichandran, V.; Agarwal, R.K. 1,3,4-Thiadiazole and its derivatives: A review on recent progress in biological activities. Chem. Biol. Drug Des. 2015, 81, 557–576. [Google Scholar] [CrossRef]
- Patel, C.N.; Georrge, J.J.; Modi, K.M.; Narechania, M.B.; Patel, D.P.; Gonzalez, F.J.; Pandya, H.A. Pharmacophore-based virtual screening of catechol-o-methyltransferase (COMT) inhibitors to combat Alzheimer’s disease. J. Biomol. Struct. Dyn. 2018, 36, 3938–3957. [Google Scholar] [CrossRef]
- Grüneberg, S.; Stubbs, M.T.; Klebe, G. Successful virtual screening for novel inhibitors of human carbonic anhydrase: Strategy and experimental confirmation. J. Med. Chem. 2002, 45, 3588–3602. [Google Scholar] [CrossRef]
- Ferdinando, G.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar]
- Abdel-Hamid, M.K.; Abdel-Hafiz, A.A.; El-koussi, N.A.; Mahfouz, N.M.; Innoceti, A.; Supuran, C.T. Design, synthesis, and docking studies of new 1,3,4-thiadiazole-2-thione derivatives with carbonic anhydrase inhibitory activity. Bioorg. Med. Chem. 2007, 15, 6975–6984. [Google Scholar] [CrossRef]
- Mills, N. ChemDraw Ultra 10.0. J. Am. Chem. Soc. 2006, 128, 13649–13650. [Google Scholar] [CrossRef]
- LigPrep; Schrödinger, LLC: New York, NY, USA, 2021.
- Seidel, T.; Ibis, G.; Bendix, F.; Wolber, G. Strategies for 3D pharmacophore-based virtual screening. Drug Discov. Today Technol. 2010, 7, e221–e228. [Google Scholar] [CrossRef] [PubMed]
- Phase; Schrödinger, LLC: New York, NY, USA, 2021.
- Zhang, W.; Koehler, K.F.; Zhang, P.; Cook, J.M. Development of a comprehensive pharmacophore model for the benzodiazepine receptor. Drug Des. Discov. 1995, 12, 193–248. [Google Scholar] [PubMed]
- Sippl, W. Receptor-based 3D QSAR analysis of estrogen receptor ligands–merging the accuracy of receptor-based alignments with the computational efficiency of ligand-based methods. J. Comput. Aided Mol. Des. 2000, 14, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Glide; Schrödinger, LLC: New York, NY, USA, 2021.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method Assessment Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Asati, V.; Bharti, S.K.; Das, D.; Kashaw, V.; Kashaw, S.K. Discovery of novel ALK2 inhibitors of pyrazolo-pyrimidines: A computational study. J. Biomol. Struct. Dyn. 2021, 1–15. [Google Scholar] [CrossRef]
- Bhole, R.P.; Bonde, C.G.; Bonde, S.C.; Chikhale, R.V.; Wavhale, R.D. Pharmacophore model and atom-based 3D quantitative structure activity relationship (QSAR) of human immunodeficiency virus-1 (HIV-1) capsid assembly inhibitors. J. Biomol. Struc. Dyn. 2021, 39, 718–727. [Google Scholar] [CrossRef]
- QikProp; Schrödinger, LLC: New York, NY, USA, 2021.
- Di, L.; Kerns, E. Drug-like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Nogara, P.A.; De-Aquino Saraiva, R.; Bueno, D.C.; Lissner, L.J.; Corta, C.L.D.; Braga, M.M.; Rosemberg, D.B.; Rocha, J.B.T. Virtual screening of acetylcholinesterase inhibitors using the Lipinski’s rule of five and ZINC databank. BioMed Res. Int. 2015, 2015, 870389. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Kumar, V.; Kundu, B.; Bhattacharya, D.; Preethy, N.; Reddy, M.P.; Talukdar, A. Ligand-based pharmacophore modeling, virtual screening and molecular docking studies for discovery of potential topoisomerase I inhibitors. Comput. Struc. Biotech. J. 2019, 17, 291–310. [Google Scholar] [CrossRef]
- Shah, U.A.; Deokar, H.S.; Kadam, S.S.; Kulkarni, V.M. Pharmacophore generation and atom-based 3D-QSAR of novel 2-(4-methylsulfonylphenyl) pyrimidines as COX-2 inhibitors. Mol. Divers. 2010, 14, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Lalit, M.; Gangwal, R.P.; Dhoke, G.V.; Damre, M.V.; Khandelwal, K.; Sangamwar, A.T. A combined pharmacophore modeling, 3D-QSAR and molecular docking study of substituted bicyclo-[3.3.0] oct-2-enes as liver receptor homolog-1 (LRH-1) agonists. J. Mol. Stru. 2013, 1049, 315–325. [Google Scholar] [CrossRef]
- Ghalla, H.; Issaoui, N.; Bardak, F.; Atac, A. Intermolecular interactions and molecular docking investigations on 4-methoxybenzaldehyde. Comput. Mat. Sci. 2018, 149, 291–300. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lamber, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Peele, K.A.; Durthi, C.P.; Srihansa, T.; Krupanidhi, S.; Ayyagari, V.S.; Babu, D.J.; Indira, M.; Reddy, A.R.; Venkateswarulu, T.C. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Inform. Med. Unlocked 2020, 19, 100345. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Zhang, W.; Xu, X. ADME evaluation in drug discovery. 6. Can oral bioavailability in humans be effectively predicted by simple molecular property-based rules? J. Chem. Inf. Model. 2007, 47, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.W.; Dress, K.R.; Edwards, M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 2009, 19, 5560–5564. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compounds | Structures | IC50 Value (µM) | pIC50 Value |
|---|---|---|---|---|
| 1 | 4a |  | 3 | 5.52 |
| 2 | 4b |  | 8.03 | 5.52 |
| 3 | 4c |  | 8.54 | 5.07 |
| 4 | 4d |  | 26.16 | 5.07 |
| 5 | 4e |  | 51.63 | 5.52 |
| 6 | 4f |  | 3.28 | 5.10 |
| 7 | 4g |  | 18.74 | 5.07 |
| 8 | 4h |  | 71.62 | 4.58 |
| 9 | 4i |  | 2.55 | 4.29 |
| 10 | 4j |  | 30.77 | 5.48 |
| 11 | 4k |  | 222.82 | 4.73 |
| 12 | 4l |  | 7.11 | 4.14 |
| 13 | 4m |  | 125 | 5.59 |
| 14 | 4n |  | 9.09 | 4.51 |
| 15 | 4o |  | 4.32 | 3.65 |
| 16 | 4p |  | 3.34 | 5.15 |
| 17 | 4q |  | 4.31 | 3.90 |
| 18 | 4r |  | 144.07 | 5.04 |
| 19 | 4s |  | 42.25 | 5.36 |
| 20 | 4t |  | 8.62 | 5.48 |
| 21 | 4u |  | 4.34 | 5.37 |
| 22 | 4v |  | 8.64 | 3.84 |
| 23 | 4w |  | 73.25 | 4.37 |
| 24 | 4x |  | 8.53 | 5.06 |
| 25 | 5a |  | 7.89 | 5.36 |
| 26 | 5b |  | 3.71 | 5.06 |
| 27 | 5c |  | 5.95 | 4.14 |
| HypoID | Survival | Site | Vector | Volume | Select | Matches | Inactive | Adjusted | BEDROC |
|---|---|---|---|---|---|---|---|---|---|
| DDDRR_1 | 5.4403 | 1 | 1 | 0.9301 | 1.9083 | 4 | 2.7979 | 2.6424 | 1 |
| DDDRR_2 | 5.4403 | 1 | 1 | 0.9301 | 1.9083 | 4 | 2.7979 | 2.6424 | 1 |
| DDDRR_3 | 5.4344 | 0.9999 | 1 | 0.9303 | 1.9021 | 4 | 2.8723 | 2.562 | 1 |
| DDDRR_4 | 5.4336 | 1 | 1 | 0.9306 | 1.901 | 4 | 2.5881 | 2.8455 | 1 |
| DDDRR_5 | 5.4233 | 1 | 1 | 0.9307 | 1.8906 | 4 | 2.5503 | 2.873 | 1 |
| ADDRR_1 | 5.2355 | 1 | 1 | 0.93 | 1.7036 | 4 | 2.8242 | 2.4114 | 1 |
| ADDRR_2 | 5.2177 | 1 | 1 | 0.9301 | 1.6857 | 4 | 2.898 | 2.3197 | 1 |
| ADDRR_3 | 5.2028 | 1 | 1 | 0.8972 | 1.7036 | 4 | 2.7787 | 2.4241 | 1 |
| ADDRR_4 | 5.1937 | 1 | 1 | 0.8979 | 1.6937 | 4 | 2.427 | 2.7667 | 1 |
| ADDRR_5 | 5.1834 | 0.9999 | 1 | 0.9301 | 1.6513 | 4 | 2.8282 | 2.3553 | 1 |
| DDRR_1 | 4.9585 | 1 | 1 | 0.9301 | 1.4264 | 4 | 2.8214 | 2.137 | 1 |
| DDRR_2 | 4.9585 | 1 | 1 | 0.9301 | 1.4264 | 4 | 2.8214 | 2.137 | 1 |
| DDRR_3 | 4.9527 | 1 | 1 | 0.9303 | 1.4204 | 4 | 2.9038 | 2.0489 | 1 |
| DDRR_4 | 4.9489 | 1 | 1 | 0.9301 | 1.4168 | 4 | 2.7901 | 2.1588 | 1 |
| DDRR_5 | 4.9488 | 1 | 1 | 0.9306 | 1.4162 | 4 | 2.6459 | 2.3029 | 1 |
| DDRR_6 | 4.9475 | 1 | 1 | 0.9303 | 1.4153 | 4 | 2.8664 | 2.0812 | 1 |
| DDRR_7 | 4.9451 | 0.9999 | 1 | 0.9303 | 1.4129 | 4 | 2.8672 | 2.0779 | 1 |
| DDRR_8 | 4.9444 | 1 | 1 | 0.9307 | 1.4117 | 4 | 2.6223 | 2.3221 | 1 |
| DDRR_9 | 4.9418 | 1 | 1 | 0.9306 | 1.4092 | 4 | 2.5853 | 2.3564 | 1 |
| DDRR_10 | 4.9388 | 1 | 1 | 0.9307 | 1.4061 | 4 | 2.5917 | 2.3471 | 1 |
| # Factors | SD | R^2 | R^2 CV | R^2 Scramble | F | P | RMSE | Q^2 | Pearson-r |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.4137 | 0.474 | 0.043 | 0.3174 | 17.1 | 0.00056 | 0.6 | 0.2794 | 0.8494 |
| 2 | 0.3767 | 0.5867 | 0.0764 | 0.4491 | 12.8 | 0.000352 | 0.51 | 0.4802 | 0.8547 |
| 3 | 0.3599 | 0.8438 | 0.8096 | 0.549 | 10.2 | 0.000438 | 0.47 | 0.7448 | 0.8023 |
| 4 | 0.3539 | 0.8757 | 0.8277 | 0.5943 | 58.3 | 0.000784 | 0.5 | 0.7888 | 0.7495 |
| S. No. | Compound | Docking Score | MMGBSA dG Bind (XPcomplex) kcal/mol |
|---|---|---|---|
| 1 | 4m | −5.217 | −72.8 |
| 2 | 4o | −4.866 | −80.17 |
| 3 | 4s | −4.729 | −74.67 |
| 4 | 4p | −4.641 | −83.2 |
| 5 | 5b | −4.635 | −76.8 |
| 6 | ZINC77699643 | −6.178 | −69.8 |
| 7 | ZINC89275054 | −5.743 | −84.17 |
| 8 | ZINC77671412 | −5.561 | −67.67 |
| 9 | ZINC70762033 | −5.535 | −82.2 |
| Compound | CNS | MW (<500) | Dipole | HBD (<5) | HBA (<10) | QPlogPo/w (≤5) | Rule of Five (≤1) | Rule of Three |
|---|---|---|---|---|---|---|---|---|
| 4a | −1 | 279.334 | 8.68 | 3 | 5 | 1.331 | 0 | 0 |
| 4b | −2 | 295.333 | 7.564 | 4 | 5.75 | 0.601 | 0 | 0 |
| 4c | −2 | 309.36 | 7.389 | 3 | 5.75 | 1.447 | 0 | 0 |
| 4d | −2 | 309.36 | 9.912 | 3 | 5.75 | 1.446 | 0 | 0 |
| 4e | −2 | 325.36 | 8.614 | 4 | 6.5 | 0.754 | 0 | 0 |
| 4f | −1 | 313.779 | 7.85 | 3 | 5 | 1.744 | 0 | 0 |
| 4g | −1 | 313.779 | 7.342 | 3 | 5 | 1.805 | 0 | 0 |
| 4h | −1 | 358.23 | 9.087 | 3 | 5 | 1.878 | 0 | 0 |
| 4i | −1 | 358.23 | 7.462 | 3 | 5 | 1.878 | 0 | 0 |
| 4j | −1 | 297.325 | 7.295 | 3 | 5 | 1.558 | 0 | 0 |
| 4k | −2 | 324.332 | 7.092 | 3 | 6 | 0.713 | 0 | 0 |
| 4l | −2 | 324.332 | 6.573 | 3 | 6 | 0.662 | 0 | 0 |
| 4m | −2 | 322.402 | 8.929 | 3 | 6 | 1.811 | 0 | 0 |
| 4n | −1 | 293.361 | 9.244 | 3 | 4.5 | 1.753 | 0 | 0 |
| 4o | −1 | 307.388 | 9.628 | 3 | 4.5 | 2.037 | 0 | 0 |
| 4p | −2 | 309.36 | 10.476 | 4 | 5.25 | 1.009 | 0 | 0 |
| 4q | −1 | 323.387 | 10.496 | 3 | 5.25 | 1.907 | 0 | 0 |
| 4r | −1 | 327.806 | 7.953 | 3 | 4.5 | 2.231 | 0 | 0 |
| 4s | −1 | 372.257 | 8.067 | 3 | 4.5 | 2.305 | 0 | 0 |
| 4t | −2 | 338.358 | 7.204 | 3 | 5.5 | 1.079 | 0 | 0 |
| 4u | −1 | 355.432 | 9.928 | 3 | 4.5 | 2.994 | 0 | 0 |
| 4v | −2 | 280.322 | 6.159 | 3 | 6.5 | 0.678 | 0 | 0 |
| 4w | −1 | 269.296 | 8.4 | 3 | 5.5 | 0.734 | 0 | 0 |
| 4x | −2 | 259.344 | 9.12 | 3 | 5 | 0.822 | 0 | 0 |
| 5a | −2 | 320.343 | 10.649 | 3 | 6 | 0.683 | 0 | 0 |
| 5b | −2 | 399.239 | 10.195 | 3 | 6 | 1.227 | 0 | 0 |
| 5c | −2 | 365.341 | 10.978 | 3 | 7 | 0.016 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Ali, A.; Warsi, M.H.; Rahman, M.A.; Ahsan, M.J.; Azam, F. An Insight into the Structural Requirements and Pharmacophore Identification of Carbonic Anhydrase Inhibitors to Combat Oxidative Stress at High Altitudes: An In-Silico Approach. Curr. Issues Mol. Biol. 2022, 44, 1027-1045. https://doi.org/10.3390/cimb44030068
Ali A, Ali A, Warsi MH, Rahman MA, Ahsan MJ, Azam F. An Insight into the Structural Requirements and Pharmacophore Identification of Carbonic Anhydrase Inhibitors to Combat Oxidative Stress at High Altitudes: An In-Silico Approach. Current Issues in Molecular Biology. 2022; 44(3):1027-1045. https://doi.org/10.3390/cimb44030068
Chicago/Turabian StyleAli, Amena, Abuzer Ali, Musarrat Husain Warsi, Mohammad Akhlaquer Rahman, Mohamed Jawed Ahsan, and Faizul Azam. 2022. "An Insight into the Structural Requirements and Pharmacophore Identification of Carbonic Anhydrase Inhibitors to Combat Oxidative Stress at High Altitudes: An In-Silico Approach" Current Issues in Molecular Biology 44, no. 3: 1027-1045. https://doi.org/10.3390/cimb44030068
APA StyleAli, A., Ali, A., Warsi, M. H., Rahman, M. A., Ahsan, M. J., & Azam, F. (2022). An Insight into the Structural Requirements and Pharmacophore Identification of Carbonic Anhydrase Inhibitors to Combat Oxidative Stress at High Altitudes: An In-Silico Approach. Current Issues in Molecular Biology, 44(3), 1027-1045. https://doi.org/10.3390/cimb44030068