
Development of Novel Therapeutics for Schizophrenia Treatment Based on a Selective Positive Allosteric Modulation of α1-Containing GABAARs—In Silico Approach

 ,
,
Abstract
:1. Introduction
2. Material and Methods
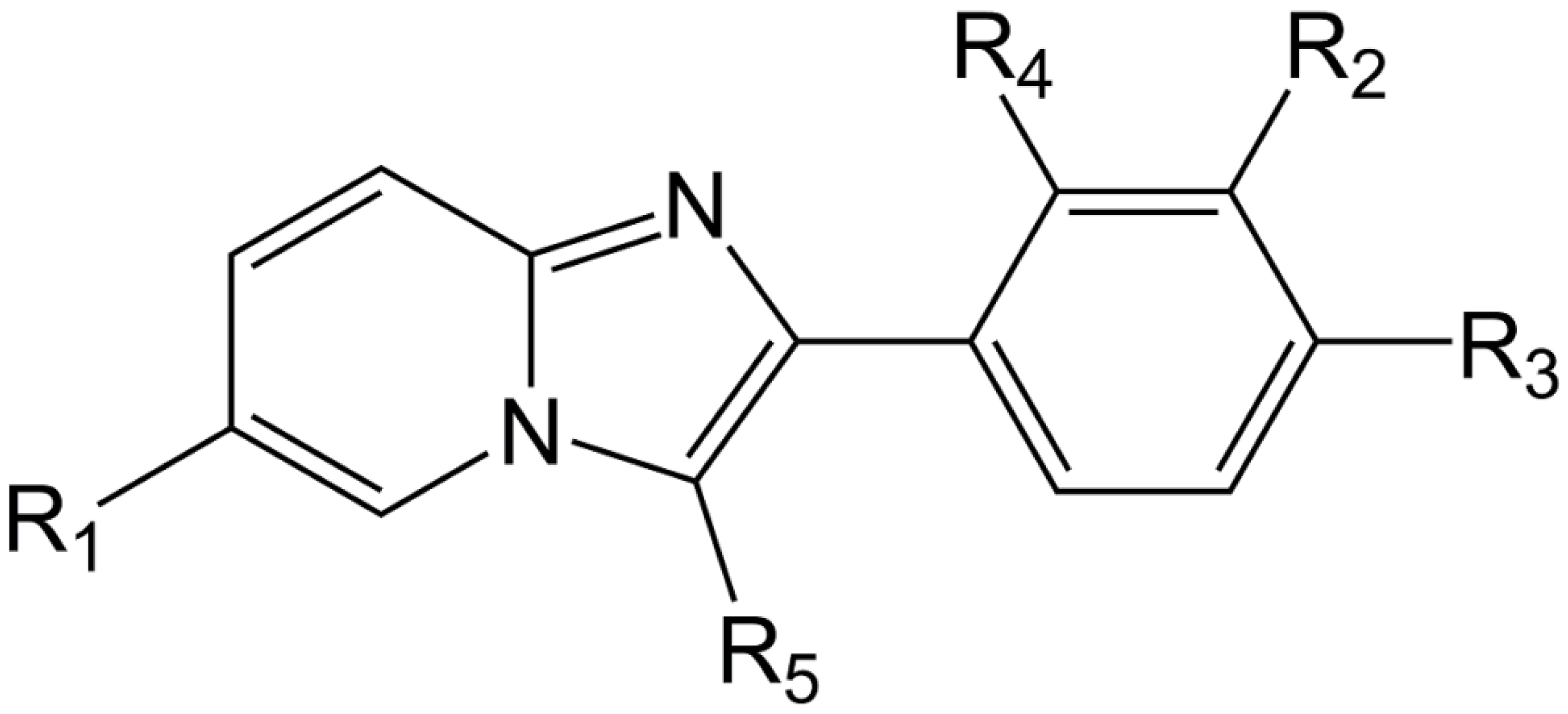
2.1. Molecules Database
2.2. The Monte Carlo Optimization Method
2.3. GA-MLR Method
2.4. QSAR Models Validation and Applicability Domain
2.5. Molecular Docking
3. Results and Discussion
3.1. The Monte Carlo Optimization Method
3.2. GA-MLR Modeling
3.3. Comparison to the Other QSAR Models
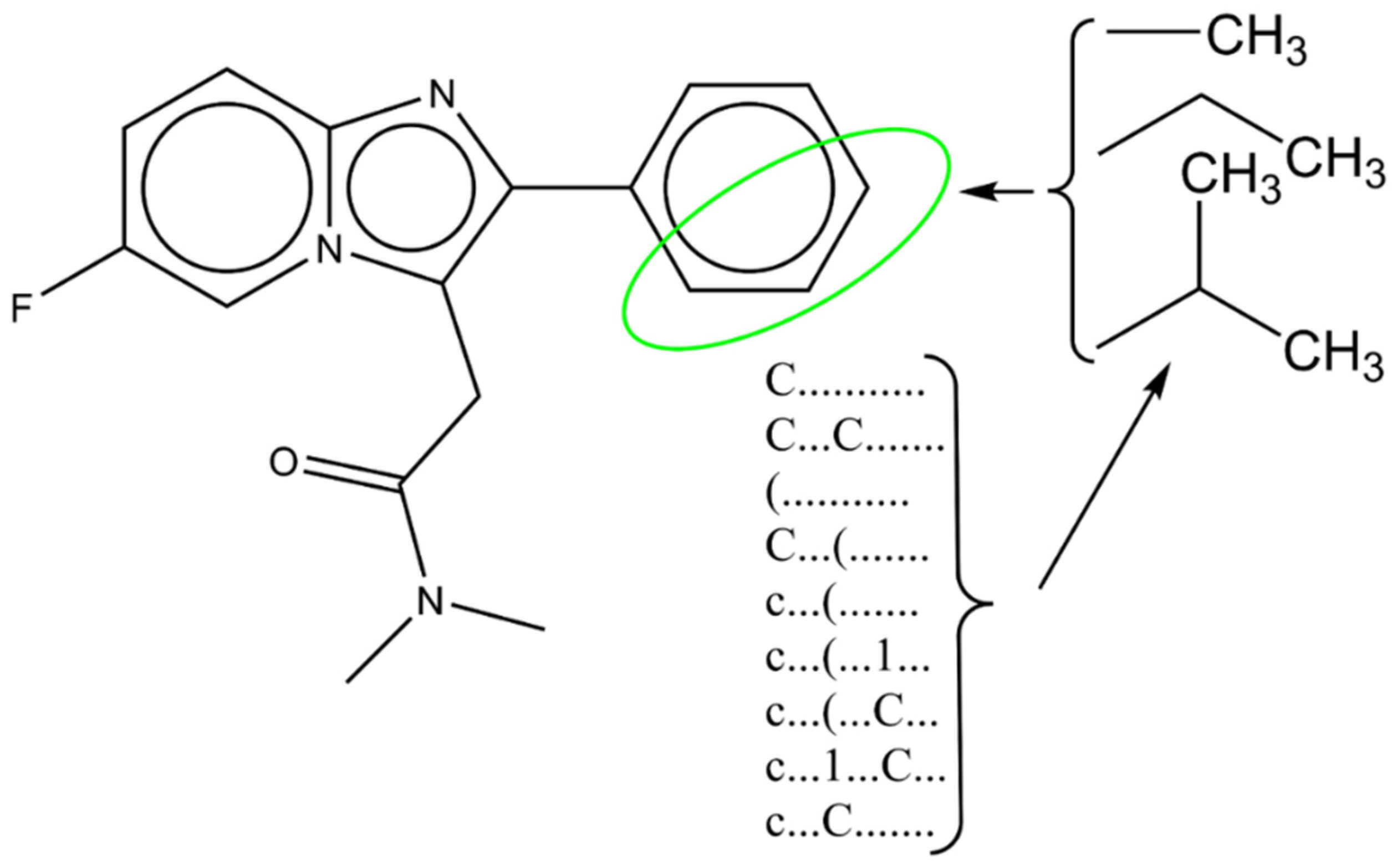
3.4. Computer-Aided Design of Novel Inhibitors
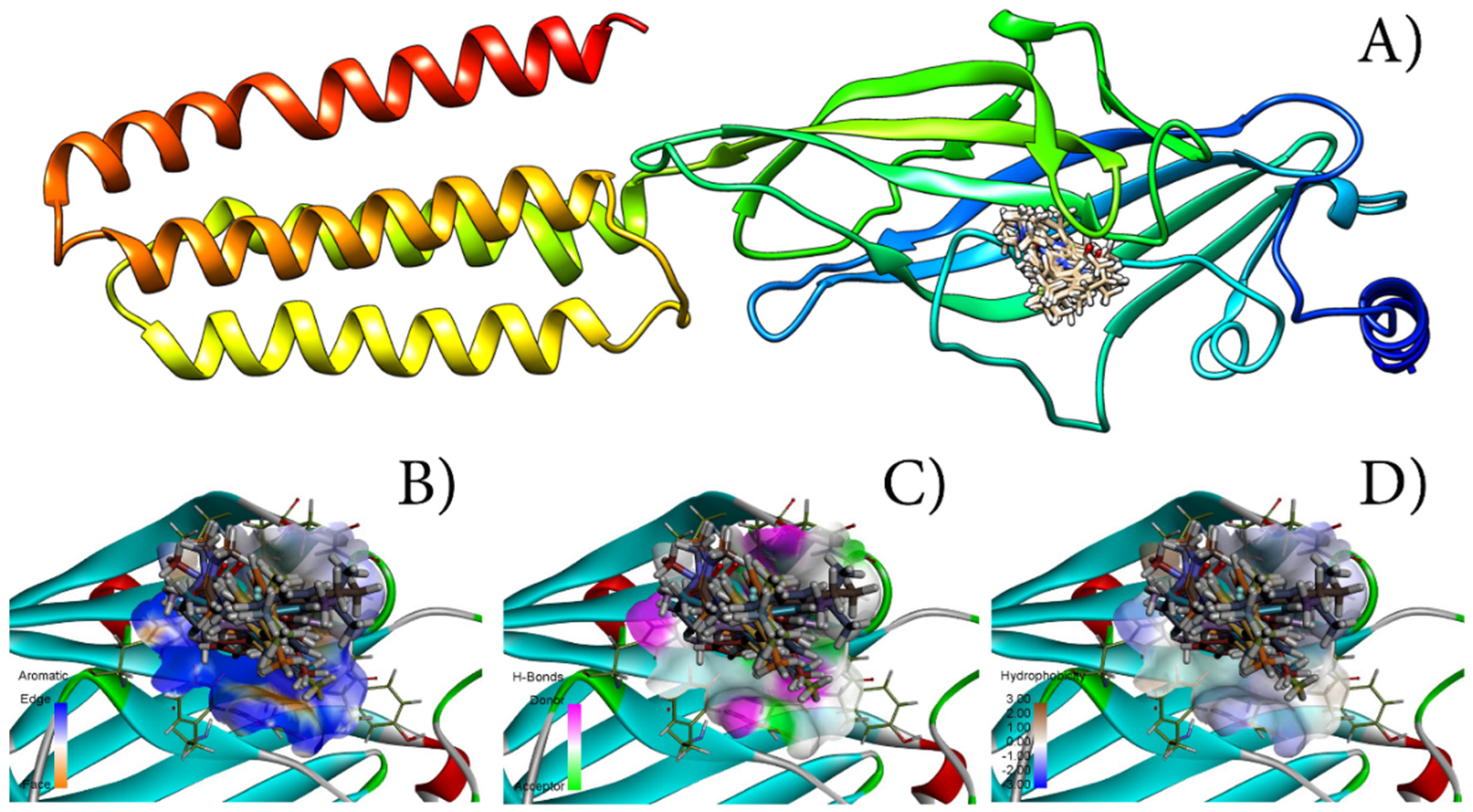
3.5. Molecular Docking
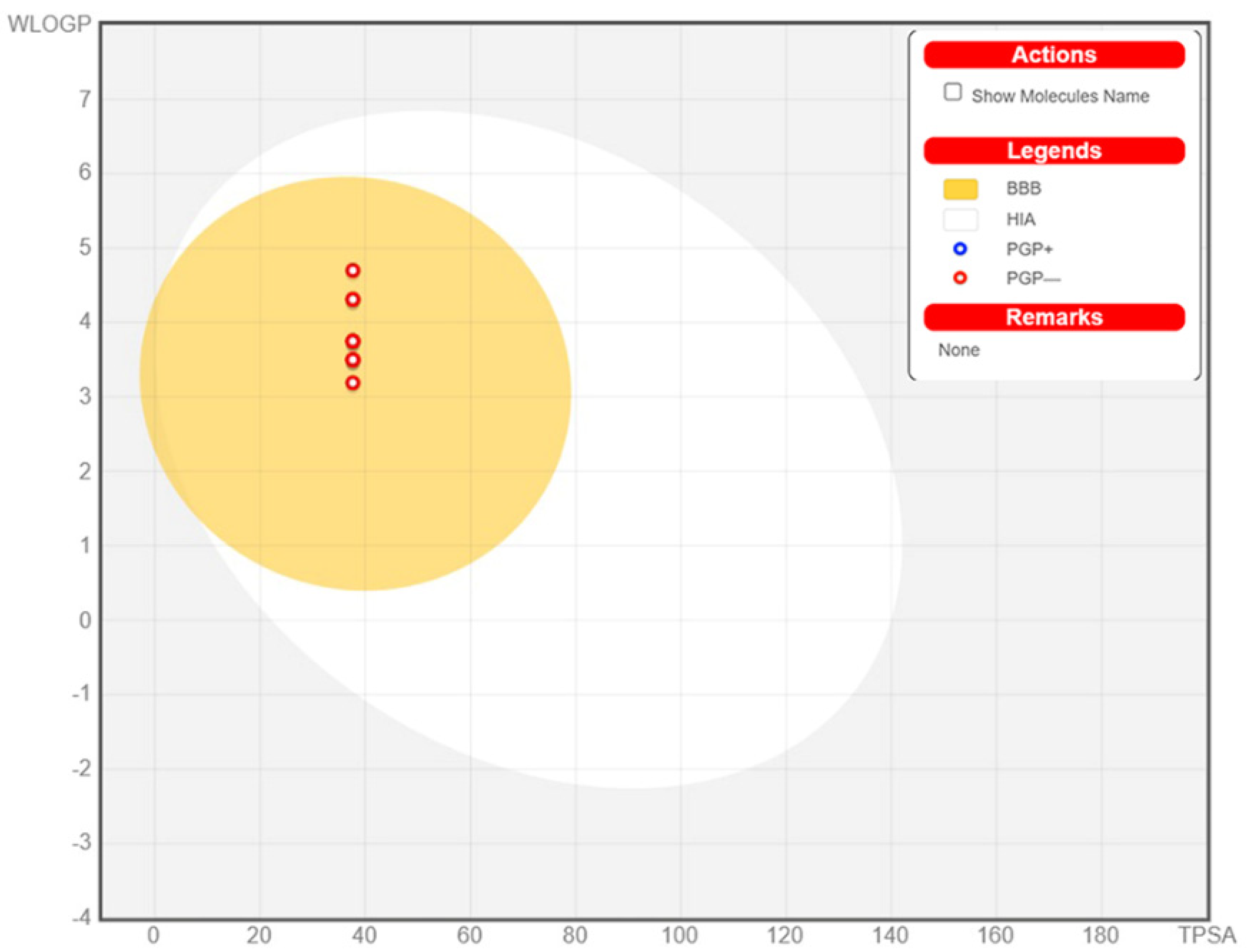
3.6. ADME Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and treatment options. Pharm. Ther. 2014, 39, 638–645. [Google Scholar]
- de Jonge, J.C.; Vinkers, C.H.; Hulshoff Pol, H.E.; Marsman, A. GABAergic Mechanisms in Schizophrenia: Linking Postmortem and In Vivo Studies. Front. Psychiatry 2017, 8, 118. [Google Scholar] [CrossRef] [Green Version]
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486. [Google Scholar] [CrossRef]
- Gogtay, N.; Vyas, N.S.; Testa, R.; Wood, S.J.; Pantelis, C. Age of onset of schizophrenia: Perspectives from structural neuroimaging studies. Schizophr. Bull. 2011, 37, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Charlson, F.J.; Ferrari, A.J.; Santomauro, D.F.; Diminic, S.; Stockings, E.; Scott, J.G.; McGrath, J.J.; Whiteford, H.A. Global Epidemiology and Burden of Schizophrenia: Findings from the Global Burden of Disease Study 2016. Schizophr. Bull. 2018, 44, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Rössler, W.; Salize, H.J.; van Os, J.; Riecher-Rössler, A. Size of burden of schizophrenia and psychotic disorders. Eur. Neuropsychopharm. 2005, 15, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W. GABAA receptor: Positive and negative allosteric modulators. Neuropharmacology 2018, 136, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Ghit, A.; Assal, D.; Al-Shami, A.S.; Hussein, D.E.E. GABAA receptors: Structure, function, pharmacology, and related disorders. J. Genet. Eng. Biotechnol. 2021, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wong, A.H.C. GABAergic inhibitory neurons as therapeutic targets for cognitive impairment in schizophrenia. Acta Pharmacol. Sin. 2018, 39, 733–753. [Google Scholar] [CrossRef] [Green Version]
- Dean, B. Understanding the pathology of schizophrenia: Recent advances from the study of the molecular architecture of postmortem CNS tissue. Postgrad. Med. J. 2002, 78, 142–148. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabeshpour, J.; Sahebkar, A.; Zirak, M.R.; Zeinali, M.; Hashemzaei, M.; Rakhshani, S.; Rakhshani, S. Computer-aided Drug Design and Drug Pharmacokinetic Prediction: A Mini-review. Curr. Pharm. Des. 2018, 24, 3014–3019. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Long, W. Current mathematical methods used in QSAR/QSPR studies. Int. J. Mol. Sci. 2009, 10, 1978–1998. [Google Scholar] [CrossRef] [PubMed]
- Pérez González, M.; Terán, C.; Saíaz-Urra, L.; Teijeira, M. Variables selection methods in QSAR: An overview. Curr. Top. Med. Chem. 2008, 8, 1606–1627. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wang, C.; Zhai, N.; Luo, X.; Liu, G.; Ju, X. In silico screening of novel α1-GABAA receptor PAMs towards schizophrenia based on combined modeling studies of imidazo [1,2-a]-pyridines. Int. J. Mol. Sci. 2021, 22, 9645. [Google Scholar] [CrossRef] [PubMed]
- Toropov, A.A.; Toropova, A.P.; Lombardo, A.; Roncaglioni, A.; Benfenati, E.; Gini, G. CORAL: Building up the model for bioconcentration factor and defining it’s applicability domain. Eur. J. Med. Chem. 2011, 46, 1400–1403. [Google Scholar] [CrossRef]
- Toropov, A.A.; Duchowicz, P.; Castro, E.A. Structure–Toxicity Relationships for Aliphatic Compounds Based on Correlation Weighting of Local Graph Invariants. Int. J. Mol. Sci. 2003, 4, 272–283. [Google Scholar] [CrossRef]
- Veselinović, A.M.; Veselinović, J.B.; Živković, J.V.; Nikolić, G.M. Application of SMILES Notation Based Optimal Descriptors in Drug Discovery and Design. Curr. Top. Med. Chem. 2015, 15, 1768–1779. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, M.; Zlatanovic, M.; Zlatanovic, N.; Golubović, M.; Veselinović, A.M. The Application of the Combination of Monte Carlo Optimization Method based QSAR Modeling and Molecular Docking in Drug Design and Development. Mini-Rev. Med. Chem. 2020, 20, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comput. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Ojha, P.K.; Roy, K. Comparative QSARs for antimalarial endochins: Importance of descriptor-thinning and noise reduction prior to feature selection. Chemometr. Intell. Lab. 2011, 109, 146–161. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Roy, P.P.; Leonard, J.T.; Roy, K. Exploring the impact of size of training sets for the development of predictive QSAR models. Chemometr. Intell. Lab. 2008, 90, 31–42. [Google Scholar] [CrossRef]
- Ojha, P.K.; Mitra, I.; Das, R.N.; Roy, K. Further exploring rm2 metrics for validation of QSPR models. Chemometr. Intell. Lab. 2011, 107, 194–205. [Google Scholar] [CrossRef]
- Roy, K.; Das, R.N.; Ambure, P.; Aher, R.B. Be aware of error measures. Further studies on validation of predictive QSAR models. Chemometr. Intell. Lab. 2016, 152, 18–33. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A. The index of ideality of correlation: A criterion of predictability of QSAR models for skin permeability? Sci. Total Environ. 2017, 586, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, D.; Mangiatordi, G.F.; Catto, M.; Carotti, A.; Nicolotti, O. Applicability Domain for QSAR Models: Where Theory Meets Reality. IJQSPR 2016, 1, 45–63. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, A.; Sindhu, J. In silico design of diacylglycerol acyltransferase-1 (DGAT1) inhibitors based on SMILES descriptors using Monte-Carlo method. SAR QSAR Environ. Res. 2019, 30, 525–541. [Google Scholar] [CrossRef]
- Chauhan, M.S.; Kumar, P.; Kumar, A. Development of prediction model for fructose- 1,6- bisphosphatase inhibitors using the Monte Carlo method. SAR QSAR Environ. Res. 2019, 30, 145–159. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2017, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
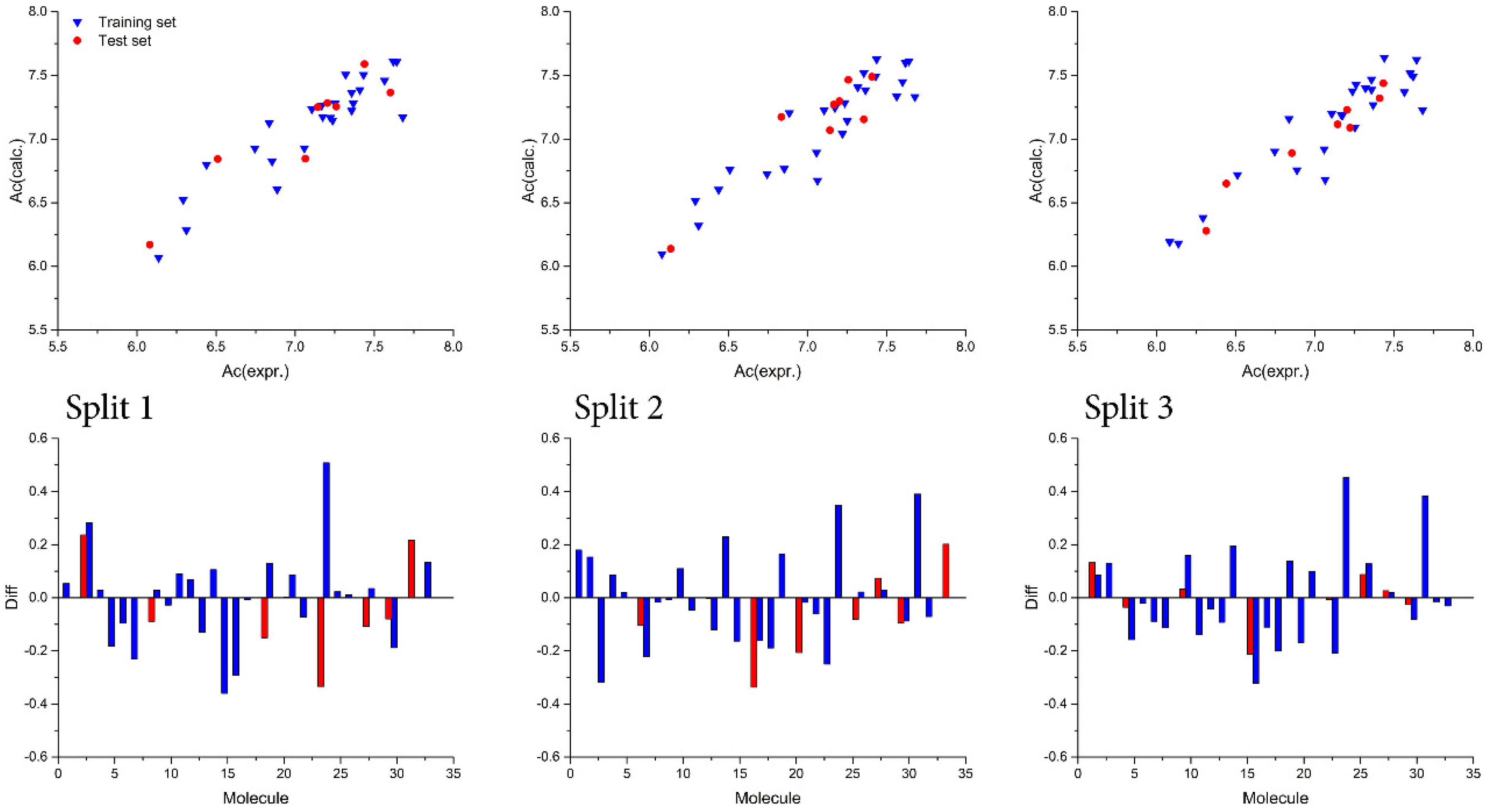
| Run | Training Set | Test Set | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r2 | CCC | IIC | q2 | s | MAE | F | r2 | CCC | IIC | q2 | s | MAE | F | ||
| Split 1 | 1 | 0.8553 | 0.922 | 0.7267 | 0.8288 | 0.166 | 0.116 | 136 | 0.8468 | 0.9192 | 0.9202 | 0.7767 | 0.207 | 0.163 | 33 |
| 2 | 0.8302 | 0.9073 | 0.7159 | 0.8059 | 0.180 | 0.127 | 112 | 0.8586 | 0.9164 | 0.9266 | 0.7872 | 0.194 | 0.152 | 36 | |
| 3 | 0.8413 | 0.9138 | 0.8467 | 0.8104 | 0.174 | 0.118 | 122 | 0.8355 | 0.9141 | 0.9140 | 0.7468 | 0.209 | 0.163 | 30 | |
| Av | 0.8423 | 0.9144 | 0.7631 | 0.8150 | 0.173 | 0.120 | 123 | 0.8470 | 0.9166 | 0.9203 | 0.7702 | 0.203 | 0.159 | 33 | |
| Split 2 | 1 | 0.8518 | 0.9200 | 0.7252 | 0.8296 | 0.179 | 0.139 | 132 | 0.8523 | 0.9086 | 0.9225 | 0.6671 | 0.181 | 0.138 | 35 |
| 2 | 0.8219 | 0.9023 | 0.8369 | 0.7888 | 0.196 | 0.159 | 106 | 0.8419 | 0.9123 | 0.9175 | 0.6644 | 0.164 | 0.114 | 32 | |
| 3 | 0.8483 | 0.9179 | 0.6140 | 0.8223 | 0.181 | 0.147 | 129 | 0.8371 | 0.9118 | 0.9145 | 0.6309 | 0.171 | 0.117 | 31 | |
| Av | 0.8407 | 0.9134 | 0.7254 | 0.8136 | 0.185 | 0.148 | 122 | 0.8438 | 0.9109 | 0.9182 | 0.6541 | 0.172 | 0.123 | 33 | |
| Split 3 | 1 | 0.8411 | 0.9137 | 0.6114 | 0.8178 | 0.182 | 0.143 | 122 | 0.9479 | 0.9678 | 0.9735 | 0.8732 | 0.103 | 0.070 | 109 |
| 2 | 0.8528 | 0.9205 | 0.6157 | 0.8283 | 0.175 | 0.132 | 133 | 0.9164 | 0.9456 | 0.9573 | 0.7794 | 0.145 | 0.121 | 66 | |
| 3 | 0.8557 | 0.9223 | 0.8539 | 0.8365 | 0.174 | 0.140 | 136 | 0.9121 | 0.9530 | 0.9540 | 0.8476 | 0.131 | 0.104 | 62 | |
| Av | 0.8499 | 0.9880 | 0.6937 | 0.8275 | 0.177 | 0.138 | 130 | 0.9255 | 9.5555 | 0.9616 | 0.8334 | 0.126 | 0.098 | 79 | |
 SMILES notation: Fc1ccc2n(c1)c(CC(=O)N(C)C)c(n2)c1ccccc1 DCW = 105.42778 Prediction for EndPoint = 6.7559 | |||||||
| SA | CW(SA) | SA | CW(SA) | SA | CW(SA) | SA | CW(SA) |
| F........... | 0.0034 | 1........... | 0.0539 | c...(....... | 0.4537 | (...N...(... | 0.0812 |
| c........... | 0.0736 | c........... | 0.0736 | c...(....... | 0.4537 | N...(...C... | 1.0483 |
| 1........... | 0.0539 | c........... | 0.0736 | n...(....... | 0.5428 | (...C...(... | −0.5696 |
| c........... | 0.0736 | c........... | 0.0736 | n...2....... | 0.2752 | C...(...C... | 0.8183 |
| c........... | 0.0736 | c........... | 0.0736 | 2...(....... | 0.8207 | (...C...(... | −0.5696 |
| c........... | 0.0736 | c........... | 0.0736 | c...(....... | 0.4537 | c...(...C... | −0.4617 |
| 2........... | 4.4302 | 1........... | 0.0539 | c...1....... | 0.0753 | (...c...(... | 2.227 |
| n........... | 1.3439 | c...F....... | 0.2931 | c...1....... | 0.0753 | n...(...c... | 0.3335 |
| (........... | −0.9688 | c...1....... | 0.0753 | c...c....... | 0.3591 | 2...n...(... | −0.7845 |
| c........... | 0.0736 | c...1....... | 0.0753 | c...c....... | 0.3591 | n...2...(... | 0.0209 |
| 1........... | 0.0539 | c...c....... | 0.3591 | c...c....... | 0.3591 | c...(...2... | 0.3706 |
| (........... | −0.9688 | c...c....... | 0.3591 | c...c....... | 0.3591 | 1...c...(... | 1.2055 |
| c........... | 0.0736 | c...2....... | −0.5812 | c...1....... | 0.0753 | c...1...c... | 0.4915 |
| (........... | −0.9688 | n...2....... | 0.2752 | F...c...1... | −0.2005 | c...c...1... | 1.4286 |
| C........... | −0.116 | n...(....... | 0.5428 | c...1...c... | 0.4915 | c...c...c... | 0.7319 |
| C........... | −0.116 | c...(....... | 0.4537 | c...c...1... | 1.4286 | c...c...c... | 0.7319 |
| (........... | −0.9688 | c...1....... | 0.0753 | c...c...c... | 0.7319 | c...c...c... | 0.7319 |
| =........... | 1.1592 | 1...(....... | 0.3914 | c...c...2... | 1.1064 | c...c...1... | 1.4286 |
| O........... | −0.0923 | c...(....... | 0.4537 | n...2...c... | 0.3482 | Cmax.2...... | 0.8467 |
| (........... | −0.9688 | c...(....... | 0.4537 | 2...n...(... | −0.7845 | Nmax.1...... | 0.0445 |
| N........... | −0.0466 | C...(....... | 0.2144 | n...(...c... | 0.3335 | Omax.1...... | 0.0472 |
| (........... | −0.9688 | C...C....... | 0.0285 | 1...c...(... | 1.2055 | Smax.0...... | 2.741 |
| C........... | −0.116 | C...(....... | 0.2144 | c...1...(... | 0.0569 | NOSP11000000 | 0.0558 |
| (........... | −0.9688 | =...(....... | 2.1975 | c...(...1... | −0.1945 | HALO10000000 | 0.5334 |
| C........... | −0.116 | O... = ....... | 1.9829 | (...c...(... | 2.227 | BOND10000000 | −0.2316 |
| (........... | −0.9688 | O...(....... | 2.1172 | c...(...C... | −0.4617 | ++++F−−−N=== | 0.6827 |
| c........... | 0.0736 | N...(....... | 0.0575 | C...C...(... | 0.0563 | ++++F−−−O=== | 0.7066 |
| (........... | −0.9688 | N...(....... | 0.0575 | C...C...(... | 0.0563 | ++++N−−−O=== | 1.2605 |
| n........... | 1.3439 | C...(....... | 0.2144 | C...(...=... | 1.221 | ++++F−−−B2== | −0.952 |
| 2........... | 4.4302 | C...(....... | 0.2144 | O...=...(... | 0.6219 | ++++O−−−B2== | 0.2153 |
| (........... | −0.9688 | C...(....... | 0.2144 | =...O...(... | 2.0622 | ++++N−−−B2== | 1.4959 |
| c........... | 0.0736 | C...(....... | 0.2144 | O...(...N... | −0.4948 | 10011001000 | 0.3564 |
| Molecule | SMILES Notation | pKi |
|---|---|---|
| A | Fc1ccc2n(c1)c(CC(=O)N(C)C)c(n2)c1ccccc1 | 6.7559 |
| A1 | Fc1ccc2n(c1)c(CC(=O)N(C)C)c(n2)c1ccccc1C | 7.4613 |
| A2 | Fc1ccc2n(c1)c(CC(=O)N(C)C)c(n2)c1cccc(c1)C | 7.1357 |
| A3 | Cc1ccc(cc1)c1nc2n(c1CC(=O)N(C)C)cc(cc2)F | 7.5160 |
| A4 | CCc1ccccc1c1nc2n(c1CC(=O)N(C)C)cc(cc2)F | 7.1291 |
| A5 | CCc1cccc(c1)c1nc2n(c1CC(=O)N(C)C)cc(cc2)F | 7.3736 |
| A6 | CCc1ccc(cc1)c1nc2n(c1CC(=O)N(C)C)cc(cc2)F | 7.3465 |
| A7 | CCN(C(=O)Cc1c(nc2n1cc(F)cc2)c1ccccc1C(C)C)C | 8.0238 |
| A8 | Fc1ccc2n(c1)c(CC(=O)N(C)C)c(n2)c1cccc(c1)C(C)C | 7.7217 |
| A9 | Fc1ccc2n(c1)c(CC(=O)N(C)C)c(n2)c1ccc(cc1)C(C)C | 7.6353 |
| Molecule | Steric | VdW | HBond | NoHBond90 | Energy | MolDock Score | Rerank Score |
|---|---|---|---|---|---|---|---|
| A | −140.362 | −46.2960 | −6.23697 | −6.65395 | −141.363 | −139.441 | −117.842 |
| A1 | −145.212 | −12.2465 | −3.76233 | −4.03557 | −145.993 | −137.563 | −86.1434 |
| A2 | −145.305 | −44.9493 | −6.96824 | −7.94622 | −148.877 | −146.146 | −121.477 |
| A3 | −140.519 | −42.7464 | −6.89116 | −7.66588 | −143.953 | −140.793 | −117.046 |
| A4 | −151.161 | −42.7453 | −2.49702 | −2.50000 | −145.106 | −143.846 | −115.584 |
| A5 | −151.189 | −46.0394 | −3.74132 | −4.28093 | −152.537 | −151.130 | −120.893 |
| A6 | −150.538 | −21.8530 | −4.48654 | −6.94755 | −146.135 | −146.097 | −108.561 |
| A7 | −148.919 | −26.6788 | −7.14605 | −7.95491 | −152.918 | −153.347 | −117.813 |
| A8 | −157.365 | −39.4826 | −2.50000 | −2.50000 | −154.518 | −150.102 | −110.772 |
| A9 | −151.100 | −17.1298 | −4.53151 | −7.03151 | −146.090 | −148.784 | −105.663 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Đorđević, V.; Petković, M.; Živković, J.; Nikolić, G.M.; Veselinović, A.M. Development of Novel Therapeutics for Schizophrenia Treatment Based on a Selective Positive Allosteric Modulation of α1-Containing GABAARs—In Silico Approach. Curr. Issues Mol. Biol. 2022, 44, 3398-3412. https://doi.org/10.3390/cimb44080234
Đorđević V, Petković M, Živković J, Nikolić GM, Veselinović AM. Development of Novel Therapeutics for Schizophrenia Treatment Based on a Selective Positive Allosteric Modulation of α1-Containing GABAARs—In Silico Approach. Current Issues in Molecular Biology. 2022; 44(8):3398-3412. https://doi.org/10.3390/cimb44080234
Chicago/Turabian StyleĐorđević, Vladimir, Milan Petković, Jelena Živković, Goran M. Nikolić, and Aleksandar M. Veselinović. 2022. "Development of Novel Therapeutics for Schizophrenia Treatment Based on a Selective Positive Allosteric Modulation of α1-Containing GABAARs—In Silico Approach" Current Issues in Molecular Biology 44, no. 8: 3398-3412. https://doi.org/10.3390/cimb44080234
APA StyleĐorđević, V., Petković, M., Živković, J., Nikolić, G. M., & Veselinović, A. M. (2022). Development of Novel Therapeutics for Schizophrenia Treatment Based on a Selective Positive Allosteric Modulation of α1-Containing GABAARs—In Silico Approach. Current Issues in Molecular Biology, 44(8), 3398-3412. https://doi.org/10.3390/cimb44080234