
Bridging PCR: An Efficient and Reliable Scheme Implemented for Genome-Walking


Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic DNA
2.2. Primer Design
2.3. Bridging PCR Procedure
2.4. DNA Manipulation and Sequencing
3. Results
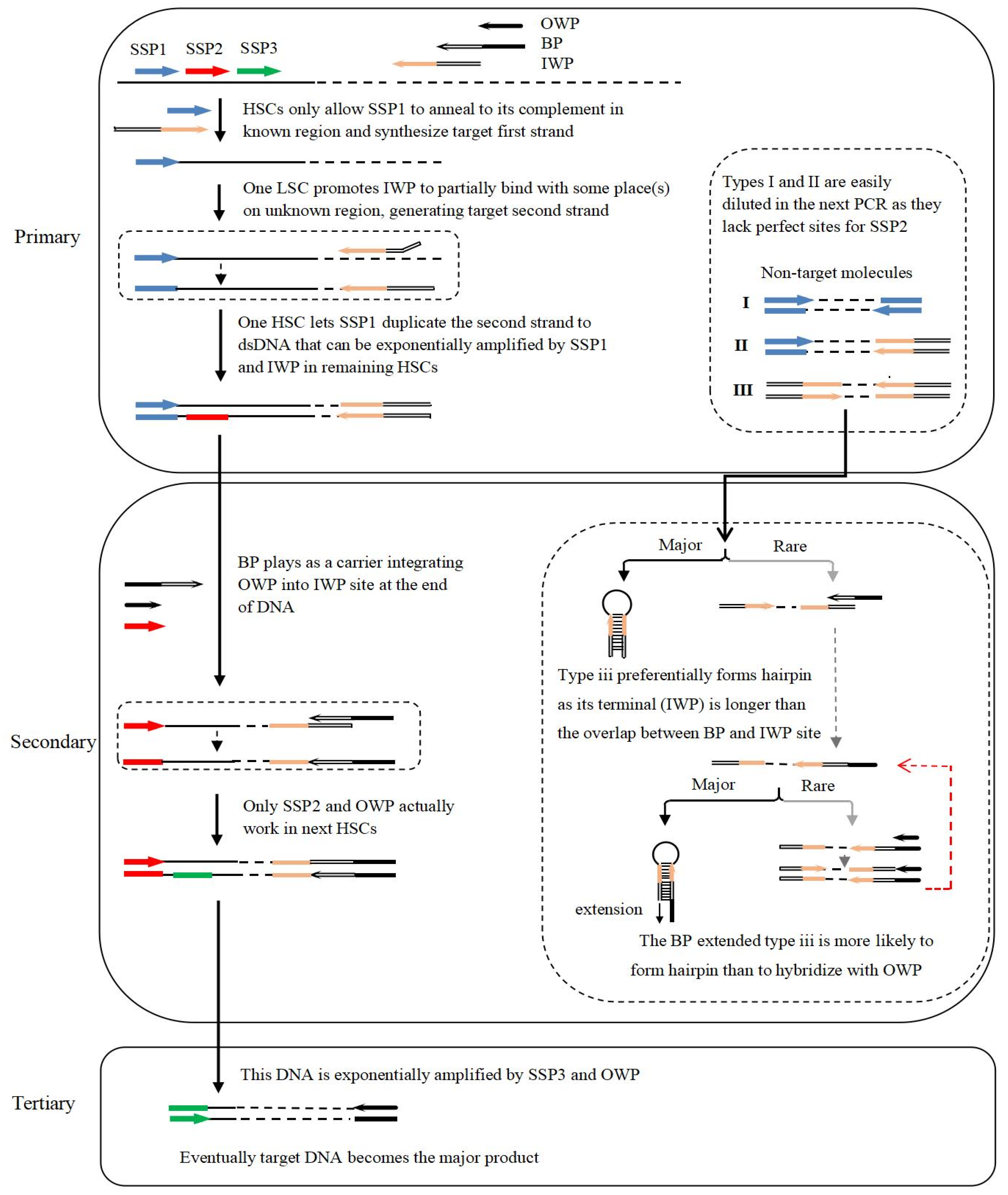
3.1. Rationale and Process of Bridging PCR
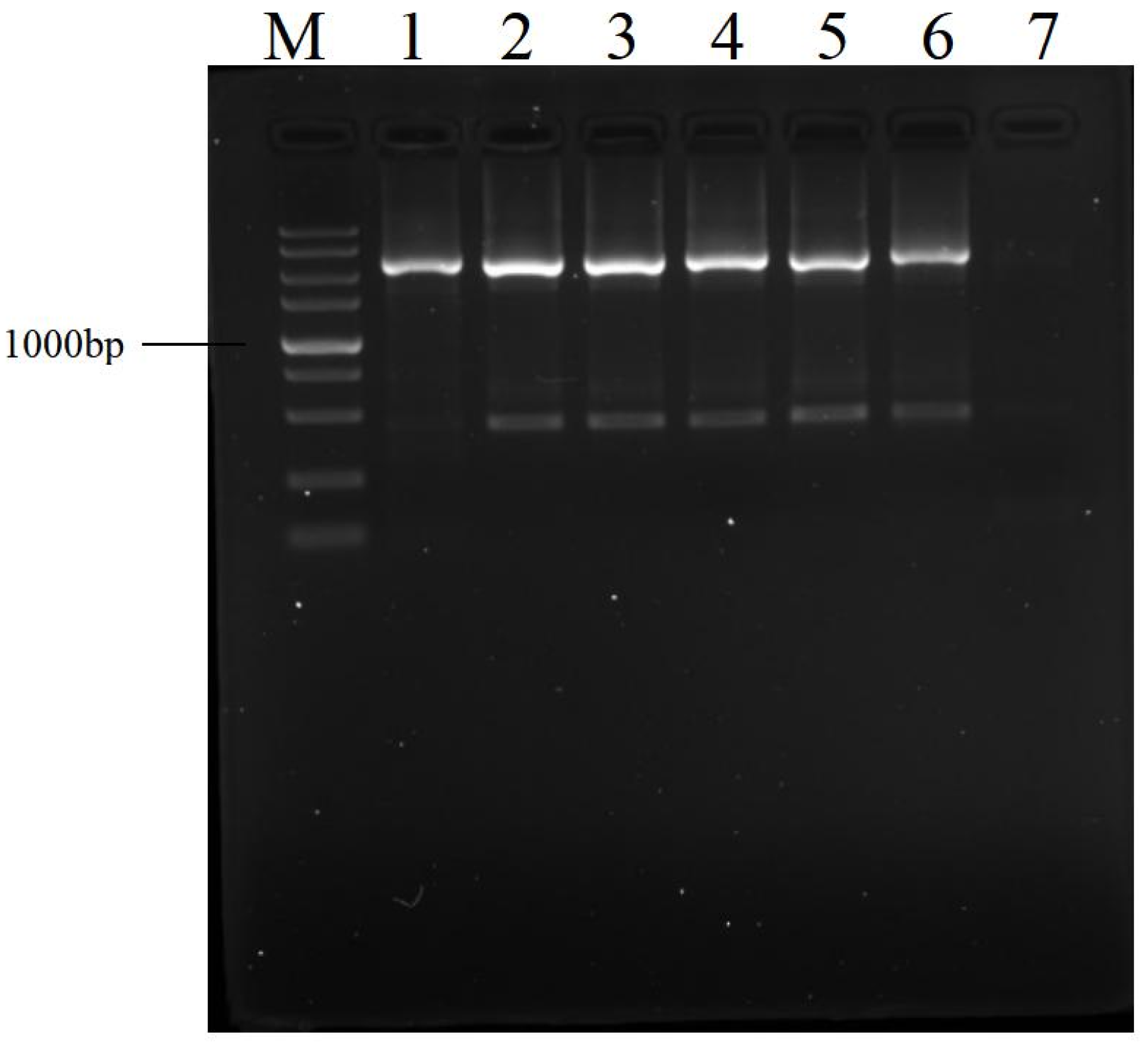
3.2. Effects of Dilution of Amplification Product on Next PCR
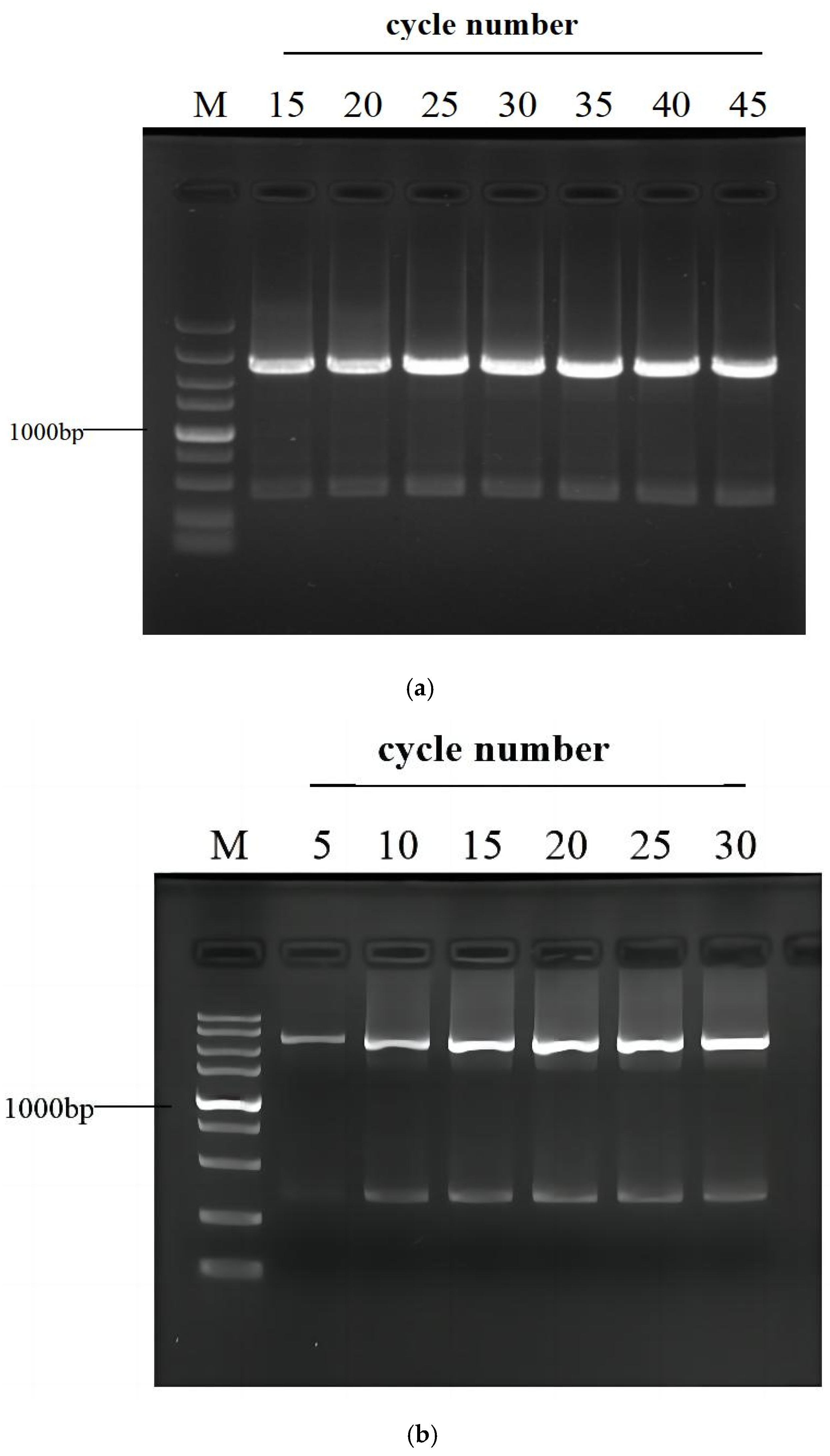
3.3. Effects of PCR Cycle Number
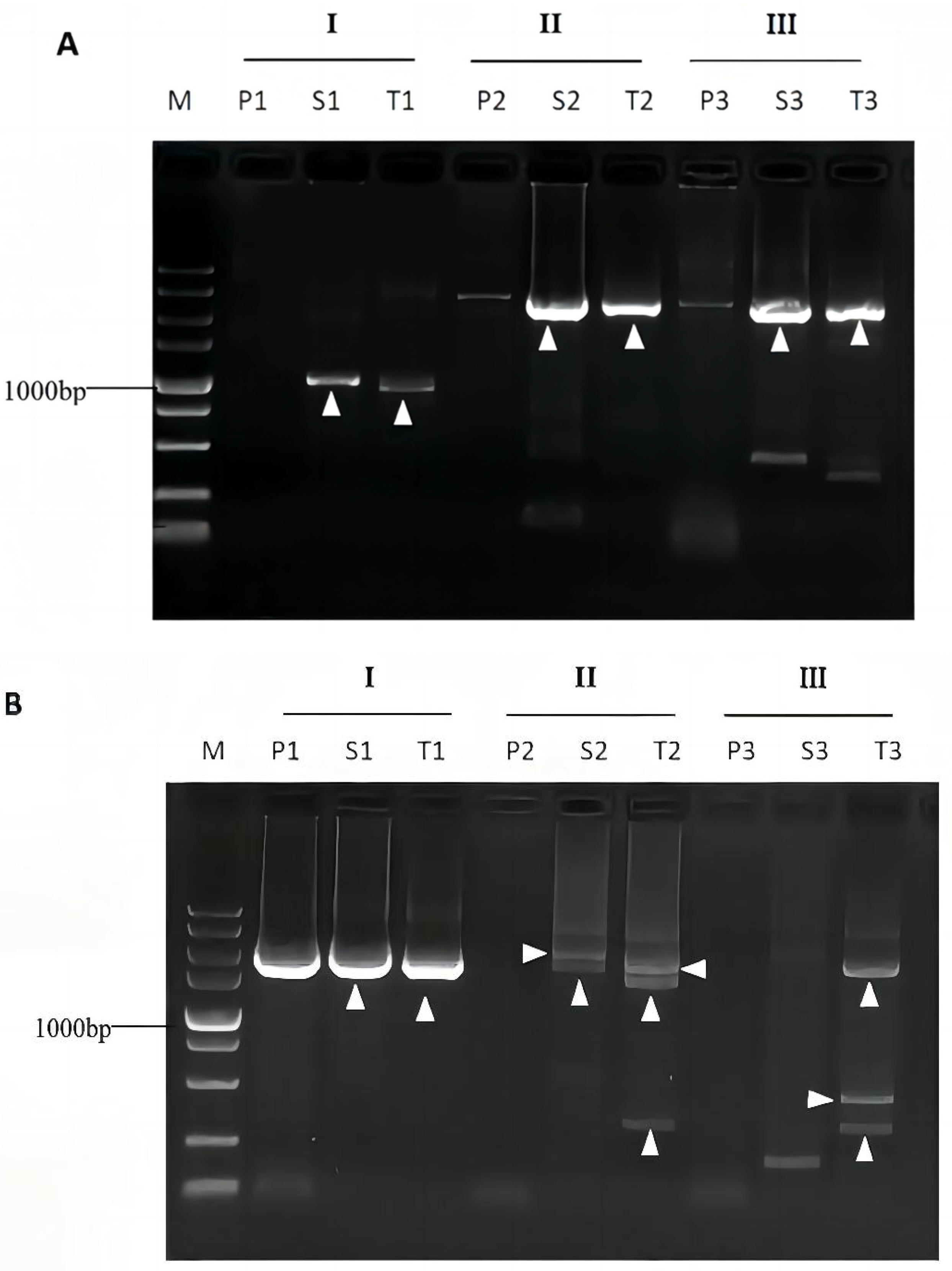
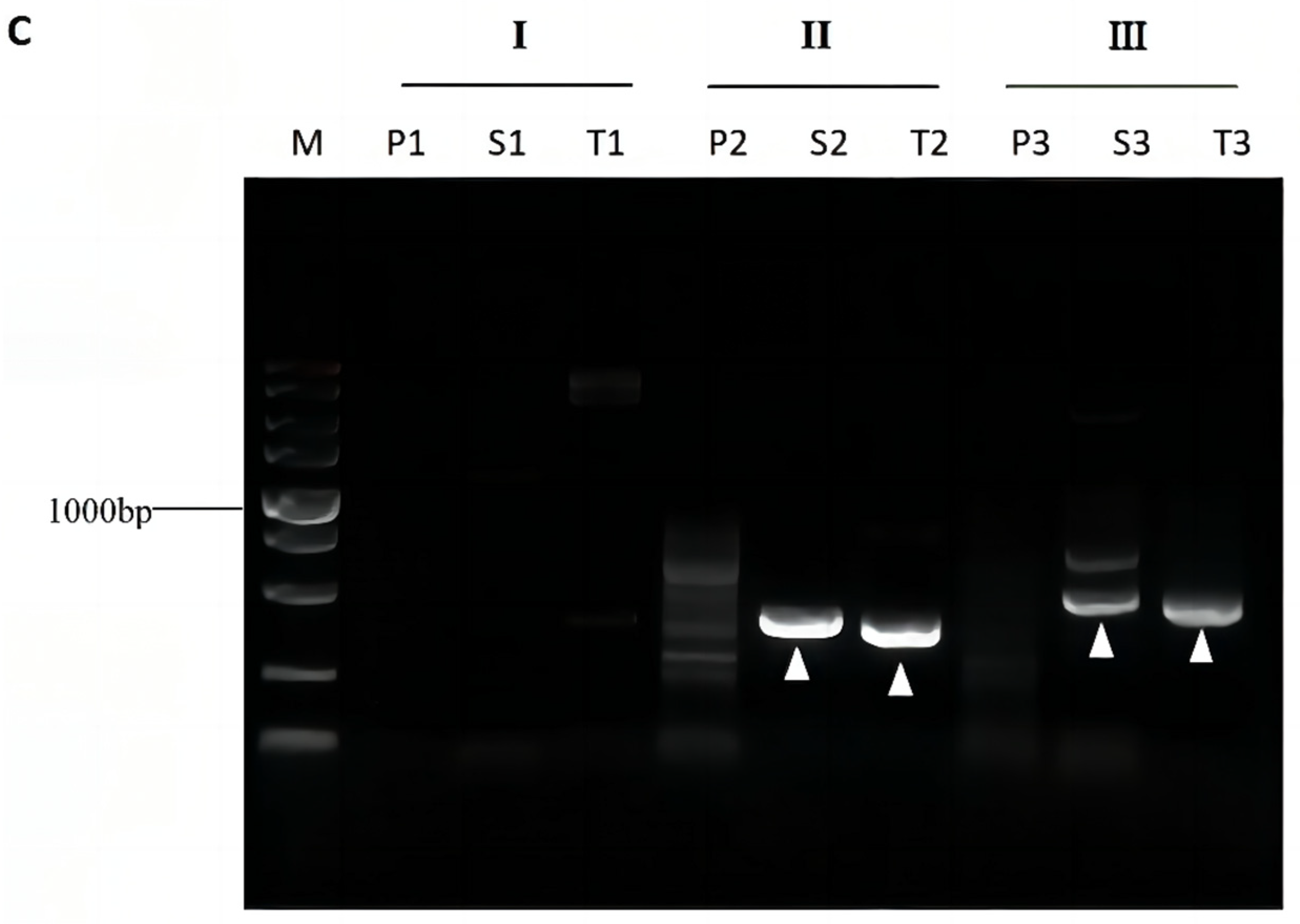
3.4. Validation of Bridging PCR
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leoni, C.; Volpicella, M.; De Leo, F.; Gallerani, R.; Ceci, L.R. Genome walking in eukaryotes. FEBS J. 2011, 278, 3953–3977. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jia, M.; Li, Z.; Liu, X.; Sun, T.; Pei, J.; Wei, C.; Lin, Z.; Li, H. Wristwatch PCR: A versatile and efficient genome walking strategy. Front. Bioeng. Biotechnol. 2022, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Kotik, M. Novel genes retrieved from environmental DNA by polymerase chain reaction: Current genome-walking techniques for future metagenome applications. J. Biotechnol. 2009, 144, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Myrick, K.V.; Gelbart, W.M. A modified universal fast walking method for single-tube transposon mapping. Nat. Protoc. 2007, 2, 1556–1563. [Google Scholar] [CrossRef]
- Fraiture, M.A.; Papazova, N.; Roosens, N.H.C. DNA walking strategy to identify unauthorized genetically modified bacteria in microbial fermentation products. Int. J. Food Microbiol. 2021, 337, 108913. [Google Scholar] [CrossRef]
- Gibaud, A.; Vogt, N.; Brison, O.; Debatisse, M.; Malfoy, B. Characterization at nucleotide resolution of the homogeneously staining region sites of insertion in two cancer cell lines. Nucleic Acids Res. 2013, 41, 8210–8219. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; He, J.; Cui, Z.; Li, S. Self-formed adaptor PCR: A simple and efficient method for chromosome walking. Appl. Environ. Microbiol. 2007, 73, 5048–5051. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ding, D.; Cao, Y.; Yu, B.; Guo, L.; Liu, X. Partially overlapping primer-based PCR for genome walking. PLoS ONE 2015, 10, e0120139. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Jia, M.; Wang, L.; Li, Z.; Lin, Z.; Wei, C.; Pei, J.; Li, H. DAR-PCR: A new tool for efficient retrieval of unknown flanking genomic DNA. AMB Express 2022, 12, 131. [Google Scholar] [CrossRef]
- Ochman, H.; Gerber, A.S.; Hartl, D.L. Genetic applications of an inverse polymerase chain reaction. Genetics 1988, 120, 621–623. [Google Scholar] [CrossRef]
- Uchiyama, T.; Watanabe, K. Improved inverse PCR scheme for metagenome walking. Biotechniques 2006, 41, 183–188. [Google Scholar] [CrossRef]
- Yan, Y.; An, C.; Li, L.; Gu, J.; Tan, G.; Chen, X. T-linker-specific ligation PCR (T-linker PCR): An advanced PCR technique for chromosome walking or for isolation of tagged DNA ends. Nucleic Acids Res. 2003, 31, e68. [Google Scholar]
- Yik, M.H.Y.; Lo, Y.T.; Lin, X.; Sun, W.; Chan, T.F.; Shaw, P.C. Authentication of Hedyotis products by adaptor ligation-mediated PCR and metabarcoding. J. Pharmaceut. Biomed. 2021, 196, 113920. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Whittier, R.F. Thermal asymmetric interlaced PCR: Automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 1995, 25, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, W.; Feng, Z.; Hong, Z. A low degenerate primer pool improved the efficiency of high-efficiency thermal asymmetric interlaced PCR to amplify T-DNA flanking sequences in Arabidopsis thaliana. 3 Biotech. 2017, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Gao, Y.; Shi, M.; Zhang, X.; He, S.; Chen, Z.; An, C. SiteFinding-PCR: A simple and efficient PCR method for chromosome walking. Nucleic Acids Res. 2005, 33, e122. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.B.; Lin, X.J.; Chen, J.C. Linear and exponential TAIL-PCR: A method for efficient and quick amplification of flanking sequences adjacent to Tn5 transposon insertion sites. AMB Express 2017, 7, 195. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Sun, T.; Wang, L.; Pan, Z.; Guo, X.; Li, H. Fusion primer driven racket PCR: A novel tool for genome walking. Front. Genet. 2022, 13, 969840. [Google Scholar] [CrossRef]
- Chang, K.; Wang, Q.; Shi, X.; Wang, S.; Wu, H.; Nie, L.; Li, H. Stepwise partially overlapping primer-based PCR for genome walking. AMB Express 2018, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Chang, K.; Ding, G.; Wu, H.; Chen, Y.; Jia, M.; Liu, X.; Wang, S.; Jin, Y.; Pan, H.; et al. Genomic insights into a robust gamma-aminobutyric acid-producer Lactobacillus brevis CD0817. AMB Express 2019, 9, 72. [Google Scholar] [CrossRef]
- Jia, M.; Zhu, Y.; Wang, L.; Sun, T.; Pan, H.; Li, H. pH auto-sustain-based fermentation supports efficient gamma-aminobutyric acid production by Lactobacillus brevis CD0817. Fermentation 2022, 8, 208. [Google Scholar] [CrossRef]
- Li, H.; Sun, T.; Jia, M.; Wang, L.; Wei, C.; Pei, J.; Lin, Z.; Wang, S. Production of gamma-aminobutyric acid by Levilactobacillus brevis CD0817 by coupling fermentation with self-buffered whole-cell catalysis. Fermentation 2022, 8, 321. [Google Scholar] [CrossRef]
- Siebert, P.D.; Chenchik, A.; Kellogg, D.E.; Lukyanov, K.A.; Lukyanov, S.A. An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res. 1995, 23, 1087–1088. [Google Scholar] [CrossRef] [PubMed]
- Chenchik, A.; Diachenko, L.; Moqadam, F.; Tarabykin, V.; Lukyanov, S.; Siebert, P.D. Full-length cDNA cloning and determination of mRNA 5′ and 3′ ends by amplification of adaptor-ligated cDNA. Biotechniques 1996, 21, 526–534. [Google Scholar] [CrossRef]
- Wei, C.; Lin, Z.; Pei, J.; Pan, H.; Li, H. Semi-site-specific primer PCR: A simple but reliable genome-walking tool. Curr. Issues Mol. Biol. 2023; accepted. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary PCR | Secondary PCR | Tertiary PCR | |
|---|---|---|---|
| Walker primer set | IWP1: GTCGTAGTCATGTATCGTCCTAGTCATCTGCTTGTTCGTCAGTCAGCGTC | BP: CAGTCAGTCTCAGCTAGTCAGTGTCGTCGTAGTCATGTATCGTCCTAGTC OWP: CAGTCAGTCTCAGCTAGTCAGTGTC | OWP: CAGTCAGTCTCAGCTAGTCAGTGTC |
| IWP2: GTCGTAGTCATGTATCGTCCTAGTCTCAGTCAGTCAGTTGCAGTCAGTCT | |||
| IWP3: GTCGTAGTCATGTATCGTCCTAGTCATCCAGAACAGTCGATTGGTTCAGC | |||
| gadA SSP set | SSP1: TCCAAGAATCATCCGCAATCGTCA | SSP2: TGGTAACATCGTCACGGTTCTTTGG | SSP3: TAGCCTTGTACCCATCTTTACCGAA |
| gadR SSP set | SSP1: TCCTTCGTTCTTGATTCCATACCCT | SSP2: CCATTTCCATAGGTTGCTCCAAGG | SSP3: GGATACTGGCTAAAATGAATTAACTCGGATAA |
| hyg SSP set | SSP1: ACGGCAATTTCGATGATGCAGCTTG | SSP2: GGGACTGTCGGGCGTACACAA | SSP3: CTGGACCGATGGCTGTGTAGAAG |
| Round of PCR | Thermal Cycling | Cycle Number |
|---|---|---|
| Primary | 95 °C, 1 min | |
| 95 °C 20 s, 65 °C 30 s, 72 °C 2 min | 5 | |
| 95 °C 20 s, 25 °C 30 s, 72 °C 2 min | 1 | |
| 95 °C 20 s, 65 °C 30 s, 72 °C 2 min | 30 | |
| 72 °C 5 min | ||
| Secondary | 95 °C, 1 min | |
| 95 °C 20 s, 65 °C 30 s, 72 °C 1.5 min | 25–45 | |
| 72 °C 5 min | ||
| Tertiary | 95 °C, 1 min | |
| 95 °C 20 s, 65 °C 30 s, 72 °C 1.5 min | 15–30 | |
| 72 °C 5 min |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Z.; Wei, C.; Pei, J.; Li, H. Bridging PCR: An Efficient and Reliable Scheme Implemented for Genome-Walking. Curr. Issues Mol. Biol. 2023, 45, 501-511. https://doi.org/10.3390/cimb45010033
Lin Z, Wei C, Pei J, Li H. Bridging PCR: An Efficient and Reliable Scheme Implemented for Genome-Walking. Current Issues in Molecular Biology. 2023; 45(1):501-511. https://doi.org/10.3390/cimb45010033
Chicago/Turabian StyleLin, Zhiyu, Cheng Wei, Jinfeng Pei, and Haixing Li. 2023. "Bridging PCR: An Efficient and Reliable Scheme Implemented for Genome-Walking" Current Issues in Molecular Biology 45, no. 1: 501-511. https://doi.org/10.3390/cimb45010033
APA StyleLin, Z., Wei, C., Pei, J., & Li, H. (2023). Bridging PCR: An Efficient and Reliable Scheme Implemented for Genome-Walking. Current Issues in Molecular Biology, 45(1), 501-511. https://doi.org/10.3390/cimb45010033