
Multiple Sclerosis: Inflammatory and Neuroglial Aspects

 , , ,
, , , 
{kind=link}
{kind=link}
Abstract
:1. Epidemiology, Etiology, Onset, Disease Course
2. Pathology: Demyelination
3. Remyelination
4. Pathogenesis: Immunologic Perspectives
4.1. Mechanisms Pertaining to T and B Lymphocytes
4.2. Mechanisms Pertaining to Innate Immunity
5. Pathogenesis: A CNS-Centered Perspective
5.1. Role of Glial Cells
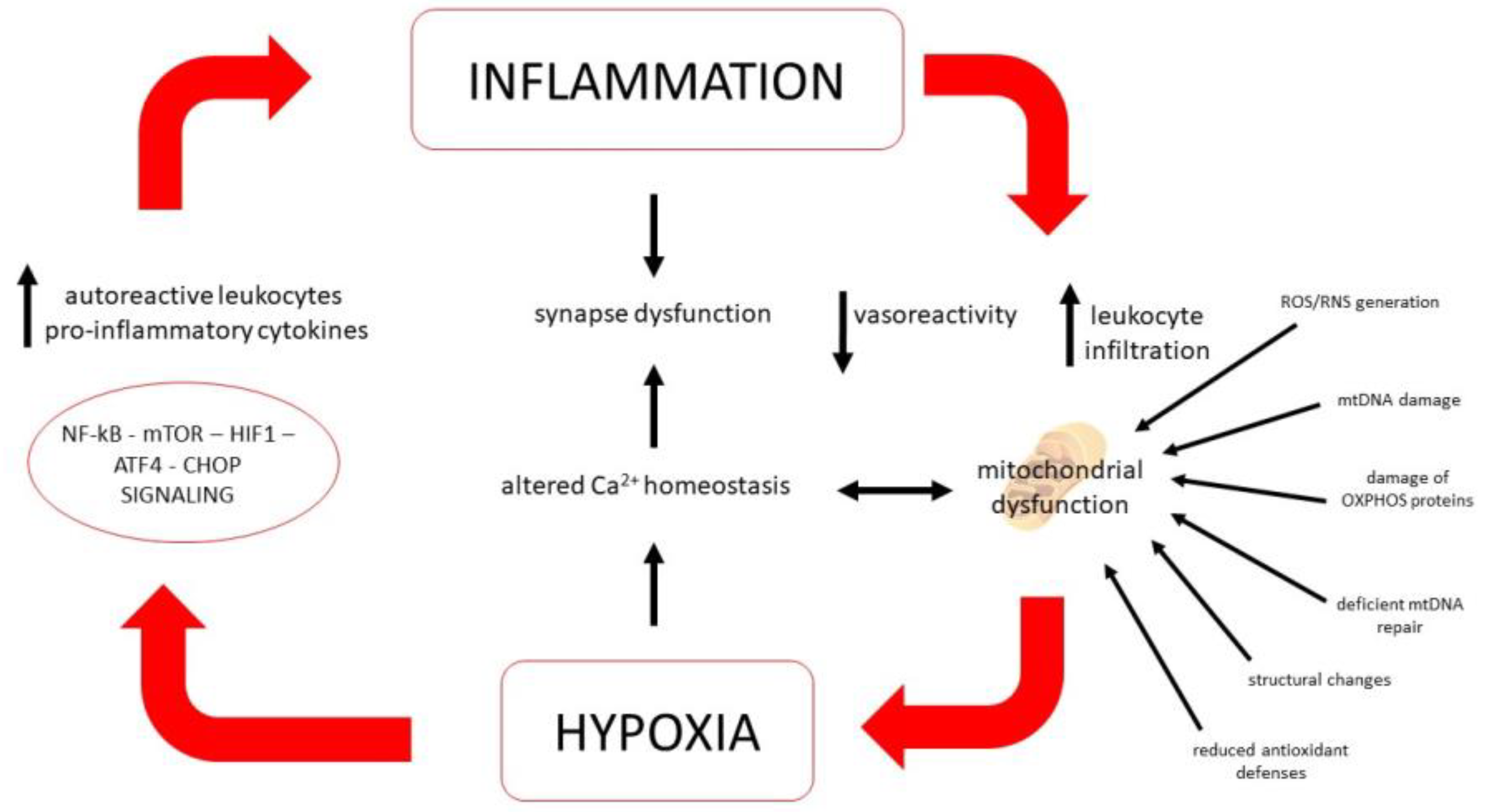
5.2. Role of Mitochondria
5.3. Ion Homeostasis and Energy Metabolism Regulation
5.4. Oxidative and Cell Stress Signaling Pathways
5.5. Synaptic Aspects
5.6. Vascular Aspects
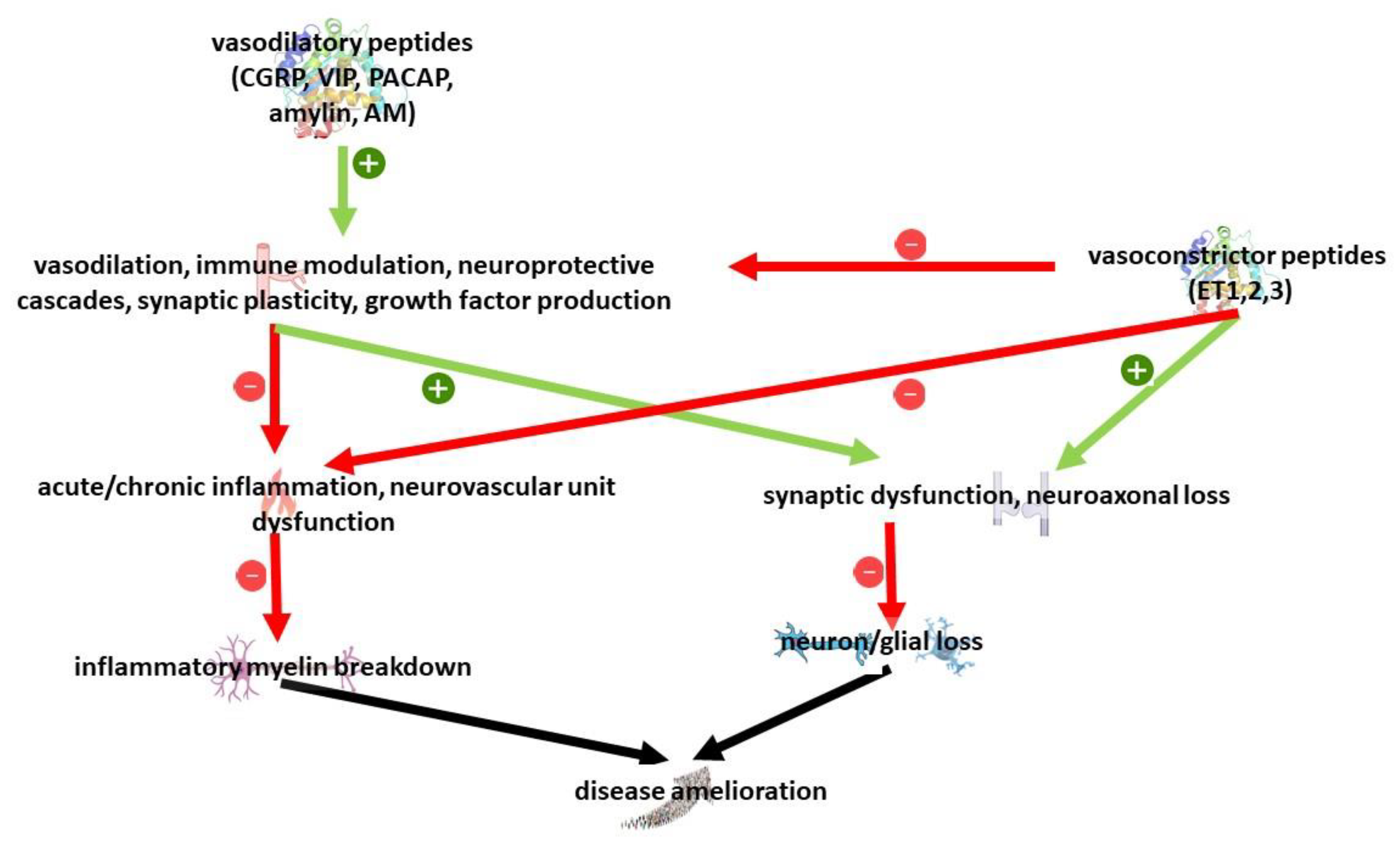
5.7. Role of Neuropeptides
6. Conclusive Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-Microarray Analysis of Multiple Sclerosis Lesions Yields New Targets Validated in Autoimmune Encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Comabella, M.; Gandhi, R. Biomarkers in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a029058. [Google Scholar] [CrossRef] [PubMed]
- Małecka, I.; Przybek-Skrzypecka, J.; Kurowska, K.; Mirowska-Guzel, D.; Członkowska, A. Clinical and Laboratory Parameters by Age for Patients Diagnosed with Multiple Sclerosis between 2000 and 2015. Neurol. Neurochir. Pol. 2021, 55, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Leray, E.; Moreau, T.; Fromont, A.; Edan, G. Epidemiology of Multiple Sclerosis. Rev. Neurol. 2016, 172, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Swank, R.L.; Lerstad, O.; Strøm, A.; Backer, J. Multiple Sclerosis in Rural Norway Its Geographic and Occupational Incidence in Relation to Nutrition. N. Engl. J. Med. 1952, 246, 722–728. [Google Scholar] [CrossRef]
- van der Mei, I.A.; Ponsonby, A.L.; Blizzard, L.; Dwyer, T. Regional Variation in Multiple Sclerosis Prevalence in Australia and Its Association with Ambient Ultraviolet Radiation. Neuroepidemiology 2001, 20, 168–174. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Giovannoni, G.; Lechner-Scott, J.; Levy, M.; Waubant, E. Multiple Sclerosis and Migration Revisited. Mult. Scler. Relat. Disord. 2019, 34, A1–A2. [Google Scholar] [CrossRef]
- Rosso, M.; Chitnis, T. Association Between Cigarette Smoking and Multiple Sclerosis. JAMA Neurol. 2020, 77, 245. [Google Scholar] [CrossRef]
- Schreiner, T.-G.; Genes, T.-M. Obesity and Multiple Sclerosis-A Multifaceted Association. J. Clin. Med. 2021, 10, 2689. [Google Scholar] [CrossRef]
- Bishir, M.; Bhat, A.; Essa, M.M.; Ekpo, O.; Ihunwo, A.O.; Veeraraghavan, V.P.; Mohan, S.K.; Mahalakshmi, A.M.; Ray, B.; Tuladhar, S.; et al. Sleep Deprivation and Neurological Disorders. BioMed Res. Int. 2020, 2020, 5764017. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, M.; de Vivo, L.; Chini, M.; Gilli, F.; Tononi, G.; Cirelli, C. Sleep Loss Promotes Astrocytic Phagocytosis and Microglial Activation in Mouse Cerebral Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 5263–5273. [Google Scholar] [CrossRef]
- Bellesi, M.; Pfister-Genskow, M.; Maret, S.; Keles, S.; Tononi, G.; Cirelli, C. Effects of Sleep and Wake on Oligodendrocytes and Their Precursors. J. Neurosci. 2013, 33, 14288–14300. [Google Scholar] [CrossRef] [PubMed]
- Brändle, S.M.; Obermeier, B.; Senel, M.; Bruder, J.; Mentele, R.; Khademi, M.; Olsson, T.; Tumani, H.; Kristoferitsch, W.; Lottspeich, F.; et al. Distinct Oligoclonal Band Antibodies in Multiple Sclerosis Recognize Ubiquitous Self-Proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 7864–7869. [Google Scholar] [CrossRef] [PubMed]
- Chapenko, S.; Millers, A.; Nora, Z.; Logina, I.; Kukaine, R.; Murovska, M. Correlation between HHV-6 Reactivation and Multiple Sclerosis Disease Activity. J. Med. Virol. 2003, 69, 111–117. [Google Scholar] [CrossRef]
- Voumvourakis, K.I.; Kitsos, D.K.; Tsiodras, S.; Petrikkos, G.; Stamboulis, E. Human Herpesvirus 6 Infection as a Trigger of Multiple Sclerosis. Mayo Clin. Proc. 2010, 85, 1023–1030. [Google Scholar] [CrossRef]
- Castellazzi, M.; Contini, C.; Tamborino, C.; Fasolo, F.; Roversi, G.; Seraceni, S.; Rizzo, R.; Baldi, E.; Tola, M.R.; Bellini, T.; et al. Epstein-Barr Virus-Specific Intrathecal Oligoclonal IgG Production in Relapsing-Remitting Multiple Sclerosis Is Limited to a Subset of Patients and Is Composed of Low-Affinity Antibodies. J. Neuroinflamm. 2014, 11, 188. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Jakimovski, D.; Ramanathan, M.; Weinstock-Guttman, B.; Zivadinov, R. The Role of Epstein-Barr Virus in Multiple Sclerosis: From Molecular Pathophysiology to in Vivo Imaging. Neural Regen. Res. 2019, 14, 373–386. [Google Scholar] [CrossRef]
- Skorstad, G.; Vandvik, B.; Vartdal, F.; Holmøy, T. MS and Clinically Isolated Syndromes: Shared Specificity but Diverging Clonal Patterns of Virus-Specific IgG Antibodies Produced in Vivo and by CSF B Cells in Vitro. Eur. J. Neurol. 2009, 16, 1124–1129. [Google Scholar] [CrossRef]
- Perron, H.; Garson, J.A.; Bedin, F.; Beseme, F.; Paranhos-Baccala, G.; Komurian-Pradel, F.; Mallet, F.; Tuke, P.W.; Voisset, C.; Blond, J.L.; et al. Molecular Identification of a Novel Retrovirus Repeatedly Isolated from Patients with Multiple Sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 1997, 94, 7583–7588. [Google Scholar] [CrossRef]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol. 2019, 10, 894. [Google Scholar] [CrossRef] [PubMed]
- Fainardi, E.; Castellazzi, M.; Tamborino, C.; Seraceni, S.; Tola, M.R.; Granieri, E.; Contini, C. Chlamydia Pneumoniae-Specific Intrathecal Oligoclonal Antibody Response Is Predominantly Detected in a Subset of Multiple Sclerosis Patients with Progressive Forms. J. Neurovirol. 2009, 15, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Cusick, M.F.; Fujinami, R.S. Role of Pathogens in Multiple Sclerosis. Int. Rev. Immunol. 2014, 33, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, J.; Patel, S. PCR for Bacterial 16S Ribosomal DNA in Multiple Sclerosis Cerebrospinal Fluid. Mult. Scler. Houndmills Basingstoke Engl. 2008, 14, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of Multiple Sclerosis: 2017 Revisions of the McDonald Criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Chard, D.T.; Ciccarelli, O. Clinically Isolated Syndromes. Lancet Neurol. 2012, 11, 157–169. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the Clinical Course of Multiple Sclerosis: The 2013 Revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Ontaneda, D.; Thompson, A.J.; Fox, R.J.; Cohen, J.A. Progressive Multiple Sclerosis: Prospects for Disease Therapy, Repair, and Restoration of Function. Lancet 2017, 389, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Bö, L.; Mörk, S.; Chang, A. Pathogenesis of Tissue Injury in MS Lesions. J. Neuroimmunol. 1999, 98, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Reynders, T.; D’haeseleer, M.; De Keyser, J.; Nagels, G.; D’hooghe, M.B. Definition, Prevalence and Predictive Factors of Benign Multiple Sclerosis. eNeurologicalSci 2017, 7, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, J.V.; Abraham, M.J.; Acevedo, J.A.G.; Rai, P.K.; Thottempudi, N.; Fang, X.; Gogia, B. Tumefactive Multiple Sclerosis (TMS): A Case Series of This Challenging Variant of MS. Mult. Scler. Relat. Disord. 2021, 48, 102699. [Google Scholar] [CrossRef] [PubMed]
- Rohani, M.; Ghourchian, S. Fulminant Multiple Sclerosis (MS). Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2011, 32, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a028936. [Google Scholar] [CrossRef] [PubMed]
- Pirko, I.; Lucchinetti, C.F.; Sriram, S.; Bakshi, R. Gray Matter Involvement in Multiple Sclerosis. Neurology 2007, 68, 634–642. [Google Scholar] [CrossRef]
- Jarius, S.; König, F.B.; Metz, I.; Ruprecht, K.; Paul, F.; Brück, W.; Wildemann, B. Pattern II and Pattern III MS Are Entities Distinct from Pattern I MS: Evidence from Cerebrospinal Fluid Analysis. J. Neuroinflamm. 2017, 14, 171. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Bruck, W.; Lassmann, H. Evidence for Pathogenic Heterogeneity in Multiple Sclerosis. Ann. Neurol. 2004, 56, 308. [Google Scholar] [CrossRef]
- Bellingacci, L.; Mancini, A.; Gaetani, L.; Tozzi, A.; Parnetti, L.; Di Filippo, M. Synaptic Dysfunction in Multiple Sclerosis: A Red Thread from Inflammation to Network Disconnection. Int. J. Mol. Sci. 2021, 22, 9753. [Google Scholar] [CrossRef]
- Lassmann, H. Mechanisms of White Matter Damage in Multiple Sclerosis. Glia 2014, 62, 1816–1830. [Google Scholar] [CrossRef]
- Carotenuto, A.; Cacciaguerra, L.; Pagani, E.; Preziosa, P.; Filippi, M.; Rocca, M.A. Glymphatic System Impairment in Multiple Sclerosis: Relation with Brain Damage and Disability. Brain J. Neurol. 2022, 145, 2785–2795. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of Ectopic B-Cell Follicles with Germinal Centers in the Meninges of Patients with Secondary Progressive Multiple Sclerosis. Brain Pathol. Zurich Switz. 2004, 14, 164–174. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical Demyelination and Diffuse White Matter Injury in Multiple Sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, J.M.; Neema, M.; Ceccarelli, A. Iron and Multiple Sclerosis. Neurobiol. Aging 2014, 35, S51–S58. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, E.; Galeel, A.A.; Ramadan, I.; Gaber, D.; Mustafa, H.; Mekky, J. Iron Deposition in Multiple Sclerosis: Overall Load or Distribution Alteration? Eur. Radiol. Exp. 2022, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B.; Cortassa, S.; Aon, M.A. Mitochondrial Ion Channels: Gatekeepers of Life and Death. Physiol. Bethesda Md 2005, 20, 303–315. [Google Scholar] [CrossRef] [PubMed]
- de Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef]
- Campbell, G.; Mahad, D.J. Mitochondrial Dysfunction and Axon Degeneration in Progressive Multiple Sclerosis. FEBS Lett. 2018, 592, 1113–1121. [Google Scholar] [CrossRef]
- Kennedy, P.G.E.; George, W.; Yu, X. The Possible Role of Neural Cell Apoptosis in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 7584. [Google Scholar] [CrossRef]
- Li, X.; Chu, Y.; Ma, R.; Dou, M.; Li, S.; Song, Y.; Lv, Y.; Zhu, L. Ferroptosis as a Mechanism of Oligodendrocyte Loss and Demyelination in Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2022, 373, 577995. [Google Scholar] [CrossRef]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of Necroptosis in Multiple Sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef]
- Prineas, J.W.; Barnard, R.O.; Kwon, E.E.; Sharer, L.R.; Cho, E.S. Multiple Sclerosis: Remyelination of Nascent Lesions. Ann. Neurol. 1993, 33, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Brück, W.; Lucchinetti, C.; Lassmann, H. Remyelination Is Extensive in a Subset of Multiple Sclerosis Patients. Brain J. Neurol. 2006, 129, 3165–3172. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Frisén, J.; Lyons, D.A. Revisiting Remyelination: Towards a Consensus on the Regeneration of CNS Myelin. Semin. Cell Dev. Biol. 2021, 116, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J.M. The Age-Related Decrease in CNS Remyelination Efficiency Is Attributable to an Impairment of Both Oligodendrocyte Progenitor Recruitment and Differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Yuen, T.J.; Silbereis, J.C.; Griveau, A.; Chang, S.M.; Daneman, R.; Fancy, S.P.J.; Zahed, H.; Maltepe, E.; Rowitch, D.H. Oligodendrocyte-Encoded HIF Function Couples Postnatal Myelination and White Matter Angiogenesis. Cell 2014, 158, 383–396. [Google Scholar] [CrossRef]
- Akay, L.A.; Effenberger, A.H.; Tsai, L.-H. Cell of All Trades: Oligodendrocyte Precursor Cells in Synaptic, Vascular, and Immune Function. Genes Dev. 2021, 35, 180–198. [Google Scholar] [CrossRef]
- Albert, M.; Antel, J.; Brück, W.; Stadelmann, C. Extensive Cortical Remyelination in Patients with Chronic Multiple Sclerosis. Brain Pathol. Zurich Switz. 2007, 17, 129–138. [Google Scholar] [CrossRef]
- Yeung, M.S.Y.; Djelloul, M.; Steiner, E.; Bernard, S.; Salehpour, M.; Possnert, G.; Brundin, L.; Frisén, J. Dynamics of Oligodendrocyte Generation in Multiple Sclerosis. Nature 2019, 566, 538–542. [Google Scholar] [CrossRef]
- Yeung, M.S.Y.; Zdunek, S.; Bergmann, O.; Bernard, S.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Brundin, L.; et al. Dynamics of Oligodendrocyte Generation and Myelination in the Human Brain. Cell 2014, 159, 766–774. [Google Scholar] [CrossRef]
- Baaklini, C.S.; Rawji, K.S.; Duncan, G.J.; Ho, M.F.S.; Plemel, J.R. Central Nervous System Remyelination: Roles of Glia and Innate Immune Cells. Front. Mol. Neurosci. 2019, 12, 225. [Google Scholar] [CrossRef]
- Clemente, D.; Ortega, M.C.; Melero-Jerez, C.; de Castro, F. The Effect of Glia-Glia Interactions on Oligodendrocyte Precursor Cell Biology during Development and in Demyelinating Diseases. Front. Cell. Neurosci. 2013, 7, 268. [Google Scholar] [CrossRef] [PubMed]
- Bramow, S.; Frischer, J.M.; Lassmann, H.; Koch-Henriksen, N.; Lucchinetti, C.F.; Sørensen, P.S.; Laursen, H. Demyelination versus Remyelination in Progressive Multiple Sclerosis. Brain 2010, 133, 2983–2998. [Google Scholar] [CrossRef] [PubMed]
- Zambonin, J.L.; Zhao, C.; Ohno, N.; Campbell, G.R.; Engeham, S.; Ziabreva, I.; Schwarz, N.; Lee, S.E.; Frischer, J.M.; Turnbull, D.M.; et al. Increased Mitochondrial Content in Remyelinated Axons: Implications for Multiple Sclerosis. Brain J. Neurol. 2011, 134, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.-M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.-È.; Rivest, S. Inefficient Clearance of Myelin Debris by Microglia Impairs Remyelinating Processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef]
- Liu, A.; Li, J.; Marin-Husstege, M.; Kageyama, R.; Fan, Y.; Gelinas, C.; Casaccia-Bonnefil, P. A Molecular Insight of Hes5-Dependent Inhibition of Myelin Gene Expression: Old Partners and New Players. EMBO J. 2006, 25, 4833–4842. [Google Scholar] [CrossRef]
- Natrajan, M.S.; de la Fuente, A.G.; Crawford, A.H.; Linehan, E.; Nuñez, V.; Johnson, K.R.; Wu, T.; Fitzgerald, D.C.; Ricote, M.; Bielekova, B.; et al. Retinoid X Receptor Activation Reverses Age-Related Deficiencies in Myelin Debris Phagocytosis and Remyelination. Brain 2015, 138, 3581–3597. [Google Scholar] [CrossRef]
- Reichert, F.; Rotshenker, S. Galectin-3 (MAC-2) Controls Microglia Phenotype Whether Amoeboid and Phagocytic or Branched and Non-Phagocytic by Regulating the Cytoskeleton. Front. Cell. Neurosci. 2019, 13, 90. [Google Scholar] [CrossRef]
- Mi, S.; Hu, B.; Hahm, K.; Luo, Y.; Kam Hui, E.S.; Yuan, Q.; Wong, W.M.; Wang, L.; Su, H.; Chu, T.-H.; et al. LINGO-1 Antagonist Promotes Spinal Cord Remyelination and Axonal Integrity in MOG-Induced Experimental Autoimmune Encephalomyelitis. Nat. Med. 2007, 13, 1228–1233. [Google Scholar] [CrossRef]
- Ahmed, Z.; Fulton, D.; Douglas, M.R. Opicinumab: Is It a Potential Treatment for Multiple Sclerosis? Ann. Transl. Med. 2020, 8, 892. [Google Scholar] [CrossRef]
- Varhaug, K.N.; Kråkenes, T.; Alme, M.N.; Vedeler, C.A.; Bindoff, L.A. Mitochondrial Complex IV Is Lost in Neurons in the Cuprizone Mouse Model. Mitochondrion 2020, 50, 58–62. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental Autoimmune Encephalomyelitis (EAE) as a Model for Multiple Sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Kawai, E.; Sato, F.; Martinez, N.E.; Minagar, A.; Al-Kofahi, M.; Yun, J.W.; Cvek, U.; Trutschl, M.; Alexander, J.S.; et al. Theiler’s Virus-Mediated Immunopathology in the CNS and Heart: Roles of Organ-Specific Cytokine and Lymphatic Responses. Front. Immunol. 2018, 9, 2870. [Google Scholar] [CrossRef] [PubMed]
- Medhasi, S.; Chantratita, N. Human Leukocyte Antigen (HLA) System: Genetics and Association with Bacterial and Viral Infections. J. Immunol. Res. 2022, 2022, 9710376. [Google Scholar] [CrossRef] [PubMed]
- Choo, S.Y. The HLA System: Genetics, Immunology, Clinical Testing, and Clinical Implications. Yonsei Med. J. 2007, 48, 11. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.C.; Lanzoni, G.; Russell, M.A.; Gerling, I.; Richardson, S.J. What the HLA-I!—Classical and Non-Classical HLA Class I and Their Potential Roles in Type 1 Diabetes. Curr. Diab. Rep. 2019, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.J.; Diehn, M.; Levy, R.; et al. Predicting HLA Class II Antigen Presentation through Integrated Deep Learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Patsopoulos, N.A. Genetics of Multiple Sclerosis: An Overview and New Directions. Cold Spring Harb. Perspect. Med. 2018, 8, a028951. [Google Scholar] [CrossRef]
- Perez-Miralles, F.C. Daclizumab in Multiple Sclerosis. Rev. Neurol. 2018, 66, 271–282. [Google Scholar]
- Lee, S.Y.; Goverman, J.M. The Influence of T Cell Ig Mucin-3 Signaling on Central Nervous System Autoimmune Disease Is Determined by the Effector Function of the Pathogenic T Cells. J. Immunol. Baltim. Md 1950 2013, 190, 4991–4999. [Google Scholar] [CrossRef]
- Huber, M.; Heink, S.; Pagenstecher, A.; Reinhard, K.; Ritter, J.; Visekruna, A.; Guralnik, A.; Bollig, N.; Jeltsch, K.; Heinemann, C.; et al. IL-17A Secretion by CD8+ T Cells Supports Th17-Mediated Autoimmune Encephalomyelitis. J. Clin. Investig. 2013, 123, 247–260. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bhan, A.K.; Gilles, F.; Kemp, M.; Kerr, C.; Weiner, H.L. Immunohistochemical Analysis of the Cellular Infiltrate in Multiple Sclerosis Lesions. Ann. Neurol. 1986, 19, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Sospedra, M.; Rosito, M.; Engelhardt, B. Current Multiple Sclerosis Treatments Have Improved Our Understanding of MS Autoimmune Pathogenesis. Eur. J. Immunol. 2016, 46, 2078–2090. [Google Scholar] [CrossRef]
- Schrempf, W.; Ziemssen, T. Glatiramer Acetate: Mechanisms of Action in Multiple Sclerosis. Autoimmun. Rev. 2007, 6, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Catalfamo, M.; Reichert-Scrivner, S.; Packer, A.; Cerna, M.; Waldmann, T.A.; McFarland, H.; Henkart, P.A.; Martin, R. Regulatory CD56 Bright Natural Killer Cells Mediate Immunomodulatory Effects of IL-2Rα-Targeted Therapy (Daclizumab) in Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5941–5946. [Google Scholar] [CrossRef]
- Sinha, S.; Boyden, A.W.; Itani, F.R.; Crawford, M.P.; Karandikar, N.J. CD8(+) T-Cells as Immune Regulators of Multiple Sclerosis. Front. Immunol. 2015, 6, 619. [Google Scholar] [CrossRef]
- Laroni, A.; Armentani, E.; Kerlero de Rosbo, N.; Ivaldi, F.; Marcenaro, E.; Sivori, S.; Gandhi, R.; Weiner, H.L.; Moretta, A.; Mancardi, G.L.; et al. Dysregulation of Regulatory CD56(Bright) NK Cells/T Cells Interactions in Multiple Sclerosis. J. Autoimmun. 2016, 72, 8–18. [Google Scholar] [CrossRef]
- Piacente, F.; Bottero, M.; Benzi, A.; Vigo, T.; Uccelli, A.; Bruzzone, S.; Ferrara, G. Neuroprotective Potential of Dendritic Cells and Sirtuins in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 4352. [Google Scholar] [CrossRef]
- Matsushita, T. Regulatory and Effector B Cells: Friends or Foes? J. Dermatol. Sci. 2019, 93, 2–7. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Xiang, W.; Xie, C.; Guan, Y. The Identification, Development and Therapeutic Potential of IL-10-Producing Regulatory B Cells in Multiple Sclerosis. J. Neuroimmunol. 2021, 354, 577520. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, X.; Cao, X.; Xu, S. T-Cell Expression of Bruton’s Tyrosine Kinase Promotes Autoreactive T-Cell Activation and Exacerbates Aplastic Anemia. Cell. Mol. Immunol. 2020, 17, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Aigrot, M.-S.; Grenningloh, R.; Stankoff, B.; Lubetzki, C.; Boschert, U.; Zalc, B. Bruton’s Tyrosine Kinase Inhibition Promotes Myelin Repair. Brain Plast. 2020, 5, 123–133. [Google Scholar] [CrossRef]
- Steinmaurer, A.; Wimmer, I.; Berger, T.; Rommer, P.S.; Sellner, J. Bruton’s Tyrosine Kinase Inhibition in the Treatment of Preclinical Models and Multiple Sclerosis. Curr. Pharm. Des. 2022, 28, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Neys, S.F.H.; Rip, J.; Hendriks, R.W.; Corneth, O.B.J. Bruton’s Tyrosine Kinase Inhibition as an Emerging Therapy in Systemic Autoimmune Disease. Drugs 2021, 81, 1605–1626. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, D.K.; Yong, V.W. Metabolic Needs of Brain-Infiltrating Leukocytes and Microglia in Multiple Sclerosis. J. Neurochem. 2021, 158, 14–24. [Google Scholar] [CrossRef] [PubMed]
- De Riccardis, L.; Rizzello, A.; Ferramosca, A.; Urso, E.; De Robertis, F.; Danieli, A.; Giudetti, A.M.; Trianni, G.; Zara, V.; Maffia, M. Bioenergetics Profile of CD4 + T Cells in Relapsing Remitting Multiple Sclerosis Subjects. J. Biotechnol. 2015, 202, 31–39. [Google Scholar] [CrossRef]
- Chen, K.; Lai, C.; Su, Y.; Bao, W.D.; Yang, L.N.; Xu, P.-P.; Zhu, L.-Q. CGAS-STING-Mediated IFN-I Response in Host Defense and Neuroinflammatory Diseases. Curr. Neuropharmacol. 2022, 20, 362–371. [Google Scholar] [CrossRef]
- Zahid, M.; Busmail, A.; Penumetcha, S.S.; Ahluwalia, S.; Irfan, R.; Khan, S.A.; Rohit Reddy, S.; Vasquez Lopez, M.E.; Mohammed, L. Tumor Necrosis Factor Alpha Blockade and Multiple Sclerosis: Exploring New Avenues. Cureus 2021, 13, e18847. [Google Scholar] [CrossRef]
- Kemanetzoglou, E.; Andreadou, E. CNS Demyelination with TNF-α Blockers. Curr. Neurol. Neurosci. Rep. 2017, 17, 36. [Google Scholar] [CrossRef]
- Piehl, F. Current and Emerging Disease-modulatory Therapies and Treatment Targets for Multiple Sclerosis. J. Intern. Med. 2021, 289, 771–791. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Uchimura, T.; Gallovic, M.D.; Thamilarasan, M.; Chou, W.-C.; Gibson, S.A.; Deng, M.; Tam, J.W.; Batty, C.J.; Williams, J.; et al. STING Agonist Mitigates Experimental Autoimmune Encephalomyelitis by Stimulating Type I IFN-Dependent and -Independent Immune-Regulatory Pathways. J. Immunol. Baltim. Md 1950 2021, 206, 2015–2028. [Google Scholar] [CrossRef] [PubMed]
- Masanneck, L.; Eichler, S.; Vogelsang, A.; Korsen, M.; Wiendl, H.; Budde, T.; Meuth, S.G. The STING-IFN-β-Dependent Axis Is Markedly Low in Patients with Relapsing-Remitting Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 9249. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is An Endogenous High-Affinity Ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Fryer, A.L.; Abdullah, A.; Taylor, J.M.; Crack, P.J. The Complexity of the CGAS-STING Pathway in CNS Pathologies. Front. Neurosci. 2021, 15, 621501. [Google Scholar] [CrossRef]
- Ma, F.; Li, B.; Yu, Y.; Iyer, S.S.; Sun, M.; Cheng, G. Positive Feedback Regulation of Type I Interferon by the Interferon-Stimulated Gene STING. EMBO Rep. 2015, 16, 202–212. [Google Scholar] [CrossRef]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor CGAS Detects Mycobacterium Tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819. [Google Scholar] [CrossRef]
- Dos Santos, A.C.; Roffê, E.; Arantes, R.M.E.; Juliano, L.; Pesquero, J.L.; Pesquero, J.B.; Bader, M.; Teixeira, M.M.; Carvalho-Tavares, J. Kinin B2 Receptor Regulates Chemokines CCL2 and CCL5 Expression and Modulates Leukocyte Recruitment and Pathology in Experimental Autoimmune Encephalomyelitis (EAE) in Mice. J. Neuroinflamm. 2008, 5, 49. [Google Scholar] [CrossRef]
- Huang, L.; Liu, M.; Jiang, W.; Ding, H.; Han, Y.; Wen, M.; Li, Y.; Liu, X.; Zeng, H. Bradykinin/Bradykinin 1 Receptor Promotes Brain Microvascular Endothelial Cell Permeability and Proinflammatory Cytokine Release by Downregulating Wnt3a. J. Biochem. Mol. Toxicol. 2022, 36, e23213. [Google Scholar] [CrossRef]
- Uzawa, A.; Mori, M.; Taniguchi, J.; Kuwabara, S. Modulation of the Kallikrein/Kinin System by the Angiotensin-Converting Enzyme Inhibitor Alleviates Experimental Autoimmune Encephalomyelitis. Clin. Exp. Immunol. 2014, 178, 245–252. [Google Scholar] [CrossRef]
- Prat, A.; Weinrib, L.; Becher, B.; Poirier, J.; Duquette, P.; Couture, R.; Antel, J.P. Bradykinin B1 Receptor Expression and Function on T Lymphocytes in Active Multiple Sclerosis. Neurology 1999, 53, 2087–2092. [Google Scholar] [CrossRef]
- Tatomir, A.; Talpos-Caia, A.; Anselmo, F.; Kruszewski, A.M.; Boodhoo, D.; Rus, V.; Rus, H. The Complement System as a Biomarker of Disease Activity and Response to Treatment in Multiple Sclerosis. Immunol. Res. 2017, 65, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Rus, H.G.; Kim, L.M.; Niculescu, F.I.; Shin, M.L. Induction of C3 Expression in Astrocytes Is Regulated by Cytokines and Newcastle Disease Virus. J. Immunol. Baltim. Md 1950 1992, 148, 928–933. [Google Scholar] [CrossRef]
- Soane, L.; Cho, H.J.; Niculescu, F.; Rus, H.; Shin, M.L. C5b-9 Terminal Complement Complex Protects Oligodendrocytes from Death by Regulating Bad through Phosphatidylinositol 3-Kinase/Akt Pathway. J. Immunol. Baltim. Md 1950 2001, 167, 2305–2311. [Google Scholar] [CrossRef] [PubMed]
- Presumey, J.; Bialas, A.R.; Carroll, M.C. Complement System in Neural Synapse Elimination in Development and Disease. Adv. Immunol. 2017, 135, 53–79. [Google Scholar] [CrossRef] [PubMed]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A Complement-Microglial Axis Drives Synapse Loss during Virus-Induced Memory Impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Aeinehband, S.; Lindblom, R.P.F.; Al Nimer, F.; Vijayaraghavan, S.; Sandholm, K.; Khademi, M.; Olsson, T.; Nilsson, B.; Ekdahl, K.N.; Darreh-Shori, T.; et al. Complement Component C3 and Butyrylcholinesterase Activity Are Associated with Neurodegeneration and Clinical Disability in Multiple Sclerosis. PLoS ONE 2015, 10, e0122048. [Google Scholar] [CrossRef]
- Ingram, G.; Hakobyan, S.; Robertson, N.P.; Morgan, B.P. Elevated Plasma C4a Levels in Multiple Sclerosis Correlate with Disease Activity. J. Neuroimmunol. 2010, 223, 124–127. [Google Scholar] [CrossRef]
- Allinovi, M.; Bellinvia, A.; Pesce, F.; Milan Manani, S.; Razzolini, L.; Brezzi, B.; Protopapa, P.; Mantero, V.; Caroti, L.; Cirami, C.L.; et al. Safety and Efficacy of Eculizumab Therapy in Multiple Sclerosis: A Case Series. Brain Sci. 2021, 11, 1341. [Google Scholar] [CrossRef]
- Huynh, J.L.; Casaccia, P. Epigenetic Mechanisms in Multiple Sclerosis: Implications for Pathogenesis and Treatment. Lancet Neurol. 2013, 12, 195–206. [Google Scholar] [CrossRef]
- Ma, N.; He, T.; Johnston, L.J.; Ma, X. Host-Microbiome Interactions: The Aryl Hydrocarbon Receptor as a Critical Node in Tryptophan Metabolites to Brain Signaling. Gut Microbes 2020, 11, 1203–1219. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.-C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I Interferons and Microbial Metabolites of Tryptophan Modulate Astrocyte Activity and Central Nervous System Inflammation via the Aryl Hydrocarbon Receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Giovannoni, G.; Knappertz, V.; Steinerman, J.R.; Tansy, A.P.; Li, T.; Krieger, S.; Uccelli, A.; Uitdehaag, B.M.J.; Montalban, X.; Hartung, H.-P.; et al. A Randomized, Placebo-Controlled, Phase 2 Trial of Laquinimod in Primary Progressive Multiple Sclerosis. Neurology 2020, 95, e1027–e1040. [Google Scholar] [CrossRef]
- Rothhammer, V.; Kenison, J.E.; Li, Z.; Tjon, E.; Takenaka, M.C.; Chao, C.-C.; Alves de Lima, K.; Borucki, D.M.; Kaye, J.; Quintana, F.J. Aryl Hydrocarbon Receptor Activation in Astrocytes by Laquinimod Ameliorates Autoimmune Inflammation in the CNS. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, 946. [Google Scholar] [CrossRef]
- Ntranos, A.; Park, H.-J.; Wentling, M.; Tolstikov, V.; Amatruda, M.; Inbar, B.; Kim-Schulze, S.; Frazier, C.; Button, J.; Kiebish, M.A.; et al. Bacterial Neurotoxic Metabolites in Multiple Sclerosis Cerebrospinal Fluid and Plasma. Brain J. Neurol. 2022, 145, 569–583. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Ruck, T.; Muraro, P.A.; Bar-Or, A.; Wiendl, H. Immune Reconstitution Therapies: Concepts for Durable Remission in Multiple Sclerosis. Nat. Rev. Neurol. 2020, 16, 56–62. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Sellebjerg, F. Pulsed Immune Reconstitution Therapy in Multiple Sclerosis. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419836913. [Google Scholar] [CrossRef]
- AlSharoqi, I.A.; Aljumah, M.; Bohlega, S.; Boz, C.; Daif, A.; El-Koussa, S.; Inshasi, J.; Kurtuncu, M.; Müller, T.; Retief, C.; et al. Immune Reconstitution Therapy or Continuous Immunosuppression for the Management of Active Relapsing-Remitting Multiple Sclerosis Patients? A Narrative Review. Neurol. Ther. 2020, 9, 55–66. [Google Scholar] [CrossRef]
- Amor, S.; Monson, N.; Sellner, J.; Van Luijn, M.M.; Van Langelaar, J.; Rijvers, L.; Smolders, J. B and T Cells Driving Multiple Sclerosis: Identity, Mechanisms and Potential Triggers. Front. Immunol. 2020, 11, 760. [Google Scholar] [CrossRef]
- Pucci, E.; Giuliani, G.; Solari, A.; Simi, S.; Minozzi, S.; Di Pietrantonj, C.; Galea, I. Natalizumab for Relapsing Remitting Multiple Sclerosis. Cochrane Database Syst. Rev. 2011, 10, CD007621. [Google Scholar] [CrossRef]
- Kapoor, R.; Ho, P.-R.; Campbell, N.; Chang, I.; Deykin, A.; Forrestal, F.; Lucas, N.; Yu, B.; Arnold, D.L.; Freedman, M.S.; et al. Effect of Natalizumab on Disease Progression in Secondary Progressive Multiple Sclerosis (ASCEND): A Phase 3, Randomised, Double-Blind, Placebo-Controlled Trial with an Open-Label Extension. Lancet Neurol. 2018, 17, 405–415. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Koch-Henriksen, N.; Petersen, T.; Ravnborg, M.; Oturai, A.; Sellebjerg, F. Recurrence or Rebound of Clinical Relapses after Discontinuation of Natalizumab Therapy in Highly Active MS Patients. J. Neurol. 2014, 261, 1170–1177. [Google Scholar] [CrossRef]
- Hartung, H.-P.; Derfuss, T.; Cree, B.A.; Sormani, M.P.; Selmaj, K.; Stutters, J.; Prados, F.; MacManus, D.; Schneble, H.-M.; Lambert, E.; et al. Efficacy and Safety of Temelimab in Multiple Sclerosis: Results of a Randomized Phase 2b and Extension Study. Mult. Scler. Houndmills Basingstoke Engl. 2022, 28, 429–440. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Blutstein, T.; Haydon, P.G. The Tripartite Synapse. In The Synapse; Elsevier: Amsterdam, The Netherlands, 2014; pp. 155–172. [Google Scholar]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite Synapses: Astrocytes Process and Control Synaptic Information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Reddy, P.H. Is Multiple Sclerosis a Mitochondrial Disease? Biochim. Biophys. Acta 2010, 1802, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Bondan, E.F.; Custódio, P.R.; Lallo, M.A.; Bentubo, H.D.L.; Graça, D.L. Ethidium Bromide-Induced Demyelination in the Sciatic Nerve of Diabetic Rats. Arq. Neuropsiquiatr. 2009, 67, 1066–1070. [Google Scholar] [CrossRef]
- Licht-Mayer, S.; Campbell, G.R.; Mehta, A.R.; McGill, K.; Symonds, A.; Al-Azki, S.; Pryce, G.; Zandee, S.; Zhao, C.; Kipp, M.; et al. Axonal Response of Mitochondria to Demyelination and Complex IV Activity within Demyelinated Axons in Experimental Models of Multiple Sclerosis. Neuropathol. Appl. Neurobiol. 2022, 49, e12851. [Google Scholar] [CrossRef]
- Sprenkle, N.T.; Sims, S.G.; Sánchez, C.L.; Meares, G.P. Endoplasmic Reticulum Stress and Inflammation in the Central Nervous System. Mol. Neurodegener. 2017, 12, 42. [Google Scholar] [CrossRef]
- Cunnea, P.; Mháille, A.N.; McQuaid, S.; Farrell, M.; McMahon, J.; FitzGerald, U. Expression Profiles of Endoplasmic Reticulum Stress-Related Molecules in Demyelinating Lesions and Multiple Sclerosis. Mult. Scler. Houndmills Basingstoke Engl. 2011, 17, 808–818. [Google Scholar] [CrossRef]
- Yu, Z.; Luo, H.; Fu, W.; Mattson, M.P. The Endoplasmic Reticulum Stress-Responsive Protein GRP78 Protects Neurons against Excitotoxicity and Apoptosis: Suppression of Oxidative Stress and Stabilization of Calcium Homeostasis. Exp. Neurol. 1999, 155, 302–314. [Google Scholar] [CrossRef]
- Blass, J.P. The Mitochondrial Spiral. An Adequate Cause of Dementia in the Alzheimer’s Syndrome. Ann. N. Y. Acad. Sci. 2000, 924, 170–183. [Google Scholar] [CrossRef]
- Blass, J.P. Brain Metabolism and Brain Disease: Is Metabolic Deficiency the Proximate Cause of Alzheimer Dementia? J. Neurosci. Res. 2001, 66, 851–856. [Google Scholar] [CrossRef]
- Camara-Lemarroy, C.R.; Metz, L.; Smith, E.E.; Dunn, J.F.; Yong, V.W. Expanding the Potential Therapeutic Options for Remote Ischemic Preconditioning: Use in Multiple Sclerosis. Front. Neurol. 2018, 9, 475. [Google Scholar] [CrossRef]
- Kang, T.M.; Hilgemann, D.W. Multiple Transport Modes of the Cardiac Na+/Ca2+ Exchanger. Nature 2004, 427, 544–548. [Google Scholar] [CrossRef]
- Hilgemann, D.W. Steady-State and Dynamic Properties of Cardiac Sodium-Calcium Exchange. Sodium-Dependent Inactivation. J. Gen. Physiol. 1992, 100, 905–932. [Google Scholar] [CrossRef]
- Inglese, M.; Madelin, G.; Oesingmann, N.; Babb, J.S.; Wu, W.; Stoeckel, B.; Herbert, J.; Johnson, G. Brain Tissue Sodium Concentration in Multiple Sclerosis: A Sodium Imaging Study at 3 Tesla. Brain J. Neurol. 2010, 133, 847–857. [Google Scholar] [CrossRef]
- Maiolino, M.; Castaldo, P.; Lariccia, V.; Piccirillo, S.; Amoroso, S.; Magi, S. Essential Role of the Na+-Ca2+ Exchanger (NCX) in Glutamate-Enhanced Cell Survival in Cardiac Cells Exposed to Hypoxia/Reoxygenation. Sci. Rep. 2017, 7, 13073. [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Lariccia, V.; Castaldo, P.; Arcangeli, S.; Nasti, A.A.; Giordano, A.; Amoroso, S. Physical and Functional Interaction of NCX1 and EAAC1 Transporters Leading to Glutamate-Enhanced ATP Production in Brain Mitochondria. PLoS ONE 2012, 7, e34015. [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Arcangeli, S.; Castaldo, P.; Nasti, A.A.; Berrino, L.; Piegari, E.; Bernardini, R.; Amoroso, S.; Lariccia, V. Glutamate-Induced ATP Synthesis: Relationship between Plasma Membrane Na+/Ca2+ Exchanger and Excitatory Amino Acid Transporters in Brain and Heart Cell Models. Mol. Pharmacol. 2013, 84, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Lariccia, V.; Amoroso, S. Calcium- and ATP-Dependent Regulation of Na/Ca Exchange Function in BHK Cells: Comparison of NCX1 and NCX3 Exchangers. Cell Calcium 2018, 73, 95–103. [Google Scholar] [CrossRef]
- Minelli, A.; Castaldo, P.; Gobbi, P.; Salucci, S.; Magi, S.; Amoroso, S. Cellular and Subcellular Localization of Na+-Ca2+ Exchanger Protein Isoforms, NCX1, NCX2, and NCX3 in Cerebral Cortex and Hippocampus of Adult Rat. Cell Calcium 2007, 41, 221–234. [Google Scholar] [CrossRef]
- Magi, S.; Castaldo, P.; Macrì, M.L.; Maiolino, M.; Matteucci, A.; Bastioli, G.; Gratteri, S.; Amoroso, S.; Lariccia, V. Intracellular Calcium Dysregulation: Implications for Alzheimer’s Disease. BioMed Res. Int. 2016, 2016, 6701324. [Google Scholar] [CrossRef]
- Stirling, D.P.; Stys, P.K. Mechanisms of Axonal Injury: Internodal Nanocomplexes and Calcium Deregulation. Trends Mol. Med. 2010, 16, 160–170. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.-Y.; Dejean, L.M. Is MPTP the Gatekeeper for Necrosis, Apoptosis, or Both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Perry, G.; Zhu, X.; Moreira, P.I.; Smith, M.A. Mitochondria: The Missing Link between Preconditioning and Neuroprotection. J. Alzheimers Dis. JAD 2010, 20 (Suppl 2), S475–S485. [Google Scholar] [CrossRef]
- Yang, R.; Dunn, J.F. Multiple Sclerosis Disease Progression: Contributions from a Hypoxia-Inflammation Cycle. Mult. Scler. Houndmills Basingstoke Engl. 2019, 25, 1715–1718. [Google Scholar] [CrossRef]
- Virgili, N.; Espinosa-Parrilla, J.F.; Mancera, P.; Pastén-Zamorano, A.; Gimeno-Bayon, J.; Rodríguez, M.J.; Mahy, N.; Pugliese, M. Oral Administration of the KATP Channel Opener Diazoxide Ameliorates Disease Progression in a Murine Model of Multiple Sclerosis. J. Neuroinflamm. 2011, 8, 149. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Yang, J.; Liu, C.; Wang, S.; Zhang, C.; Zhao, H.; Du, H.; Geng, X. Hypoxia-Inducible Factor 1α and 2α Have Beneficial Effects in Remote Ischemic Preconditioning Against Stroke by Modulating Inflammatory Responses in Aged Rats. Front. Aging Neurosci. 2020, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Merelli, A.; Rodríguez, J.C.G.; Folch, J.; Regueiro, M.R.; Camins, A.; Lazarowski, A. Understanding the Role of Hypoxia Inducible Factor During Neurodegeneration for New Therapeutics Opportunities. Curr. Neuropharmacol. 2018, 16, 1484–1498. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial Stress Induced by Continuous Stimulation under Hypoxia Rapidly Drives T Cell Exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Cirac, A.; Tsaktanis, T.; Beyer, T.; Linnerbauer, M.; Andlauer, T.; Grummel, V.; Nirschl, L.; Loesslein, L.; Quintana, F.J.; Hemmer, B.; et al. The Aryl Hydrocarbon Receptor-Dependent TGF-α/VEGF-B Ratio Correlates With Disease Subtype and Prognosis in Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1043. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Karmazyn, M.; Escobales, N. Mitochondrial Permeability Transition Pore Opening as a Promising Therapeutic Target in Cardiac Diseases. J. Pharmacol. Exp. Ther. 2009, 330, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J. Signalling Pathways in Ischaemic Postconditioning. Thromb. Haemost. 2009, 101, 626–634. [Google Scholar] [CrossRef]
- Chotiyarnwong, C.; Nair, K.; Angelini, L.; Buckley, E.; Mazza, C.; Heyes, D.; Ramiz, R.; Baster, K.; Ismail, A.; Das, J.; et al. Effect of Remote Ischaemic Preconditioning on Walking in People with Multiple Sclerosis: Double-Blind Randomised Controlled Trial. BMJ Neurol. Open 2020, 2, e000022. [Google Scholar] [CrossRef]
- Misrielal, C.; Mauthe, M.; Reggiori, F.; Eggen, B.J.L. Autophagy in Multiple Sclerosis: Two Sides of the Same Coin. Front. Cell. Neurosci. 2020, 14, 603710. [Google Scholar] [CrossRef]
- Crino, P.B. The MTOR Signalling Cascade: Paving New Roads to Cure Neurological Disease. Nat. Rev. Neurol. 2016, 12, 379–392. [Google Scholar] [CrossRef]
- Dello Russo, C.; Lisi, L.; Feinstein, D.L.; Navarra, P. MTOR Kinase, a Key Player in the Regulation of Glial Functions: Relevance for the Therapy of Multiple Sclerosis. Glia 2013, 61, 301–311. [Google Scholar] [CrossRef]
- Sanadgol, N.; Barati, M.; Houshmand, F.; Hassani, S.; Clarner, T.; Shahlaei, M.; Golab, F. Metformin Accelerates Myelin Recovery and Ameliorates Behavioral Deficits in the Animal Model of Multiple Sclerosis via Adjustment of AMPK/Nrf2/MTOR Signaling and Maintenance of Endogenous Oligodendrogenesis during Brain Self-Repairing Period. Pharmacol. Rep. PR 2020, 72, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, J.; Wang, J.; Yang, B.; He, Q.; Weng, Q. Role of DJ-1 in Immune and Inflammatory Diseases. Front. Immunol. 2020, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Hirotani, M.; Maita, C.; Niino, M.; Iguchi-Ariga, S.; Hamada, S.; Ariga, H.; Sasaki, H. Correlation between DJ-1 Levels in the Cerebrospinal Fluid and the Progression of Disabilities in Multiple Sclerosis Patients. Mult. Scler. Houndmills Basingstoke Engl. 2008, 14, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Cookson, M.R. DJ-1 Regulation of Mitochondrial Function and Autophagy through Oxidative Stress. Autophagy 2011, 7, 531–532. [Google Scholar] [CrossRef]
- Zeldich, E.; Chen, C.-D.; Avila, R.; Medicetty, S.; Abraham, C.R. The Anti-Aging Protein Klotho Enhances Remyelination Following Cuprizone-Induced Demyelination. J. Mol. Neurosci. MN 2015, 57, 185–196. [Google Scholar] [CrossRef]
- Ellidag, H.Y.; Yilmaz, N.; Kurtulus, F.; Aydin, O.; Eren, E.; Inci, A.; Dolu, S.; Ince, F.D.A.; Giray, Ö.; Yaman, A. The Three Sisters of Fate in Multiple Sclerosis: Klotho (Clotho), Fibroblast Growth Factor-23 (Lachesis), and Vitamin D (Atropos). Ann. Neurosci. 2016, 23, 155–161. [Google Scholar] [CrossRef]
- Moos, W.H.; Faller, D.V.; Glavas, I.P.; Harpp, D.N.; Kanara, I.; Mavrakis, A.N.; Pernokas, J.; Pernokas, M.; Pinkert, C.A.; Powers, W.R.; et al. Klotho Pathways, Myelination Disorders, Neurodegenerative Diseases, and Epigenetic Drugs. BioResearch Open Access 2020, 9, 94–105. [Google Scholar] [CrossRef]
- Torbus-Paluszczak, M.; Bartman, W.; Adamczyk-Sowa, M. Klotho Protein in Neurodegenerative Disorders. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2018, 39, 1677–1682. [Google Scholar] [CrossRef]
- Foolad, F.; Khodagholi, F.; Javan, M. Sirtuins in Multiple Sclerosis: The Crossroad of Neurodegeneration, Autoimmunity and Metabolism. Mult. Scler. Relat. Disord. 2019, 34, 47–58. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The Emerging Role of Nrf2 in Mitochondrial Function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging Understanding of the Mechanism of Action for Dimethyl Fumarate in the Treatment of Multiple Sclerosis. Front. Neurol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Hagen, J.T.; Neufer, P.D.; Kassai, M.; Cabot, M.C. On the Nature of Ceramide-Mitochondria Interactions—Dissection Using Comprehensive Mitochondrial Phenotyping. Cell. Signal. 2021, 78, 109838. [Google Scholar] [CrossRef] [PubMed]
- McGinley, M.P.; Cohen, J.A. Sphingosine 1-Phosphate Receptor Modulators in Multiple Sclerosis and Other Conditions. Lancet Lond. Engl. 2021, 398, 1184–1194. [Google Scholar] [CrossRef]
- De Stefano, N.; Silva, D.G.; Barnett, M.H. Effect of Fingolimod on Brain Volume Loss in Patients with Multiple Sclerosis. CNS Drugs 2017, 31, 289–305. [Google Scholar] [CrossRef]
- Darios, F.; Wasser, C.; Shakirzyanova, A.; Giniatullin, A.; Goodman, K.; Munoz-Bravo, J.L.; Raingo, J.; Jorgacevski, J.; Kreft, M.; Zorec, R.; et al. Sphingosine Facilitates SNARE Complex Assembly and Activates Synaptic Vesicle Exocytosis. Neuron 2009, 62, 683–694. [Google Scholar] [CrossRef]
- Stenovec, M.; Trkov, S.; Kreft, M.; Zorec, R. Alterations of Calcium Homoeostasis in Cultured Rat Astrocytes Evoked by Bioactive Sphingolipids. Acta Physiol. Oxf. Engl. 2014, 212, 49–61. [Google Scholar] [CrossRef]
- Trkov Bobnar, S.; Stenovec, M.; Miš, K.; Pirkmajer, S.; Zorec, R. Fingolimod Suppresses the Proinflammatory Status of Interferon-γ-Activated Cultured Rat Astrocytes. Mol. Neurobiol. 2019, 56, 5971–5986. [Google Scholar] [CrossRef]
- Mandolesi, G.; Gentile, A.; Musella, A.; Fresegna, D.; De Vito, F.; Bullitta, S.; Sepman, H.; Marfia, G.A.; Centonze, D. Synaptopathy Connects Inflammation and Neurodegeneration in Multiple Sclerosis. Nat. Rev. Neurol. 2015, 11, 711–724. [Google Scholar] [CrossRef]
- Mandolesi, G.; De Vito, F.; Musella, A.; Gentile, A.; Bullitta, S.; Fresegna, D.; Sepman, H.; Di Sanza, C.; Haji, N.; Mori, F.; et al. MiR-142-3p Is a Key Regulator of IL-1β-Dependent Synaptopathy in Neuroinflammation. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 546–561. [Google Scholar] [CrossRef]
- Weiss, S.; Mori, F.; Rossi, S.; Centonze, D. Disability in Multiple Sclerosis: When Synaptic Long-Term Potentiation Fails. Neurosci. Biobehav. Rev. 2014, 43, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Nisticò, R.; Mandolesi, G.; Piccinin, S.; Mango, D.; Kusayanagi, H.; Berretta, N.; Bergami, A.; Gentile, A.; Musella, A.; et al. Interleukin-1β Promotes Long-Term Potentiation in Patients with Multiple Sclerosis. Neuromolecular Med. 2014, 16, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, S.; Huitinga, I.; Swaab, D.F. Neurosteroid and GABA-A Receptor Alterations in Alzheimer’s Disease, Parkinson’s Disease and Multiple Sclerosis. Neuroscience 2011, 191, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Dubchenko, E.; Ivanov, A.; Spirina, N.; Smirnova, N.; Melnikov, M.; Boyko, A.; Gusev, E.; Kubatiev, A. Hyperhomocysteinemia and Endothelial Dysfunction in Multiple Sclerosis. Brain Sci. 2020, 10, 637. [Google Scholar] [CrossRef]
- Paolucci, M.; Altamura, C.; Vernieri, F. The Role of Endothelial Dysfunction in the Pathophysiology and Cerebrovascular Effects of Migraine: A Narrative Review. J. Clin. Neurol. Seoul Korea 2021, 17, 164–175. [Google Scholar] [CrossRef]
- D’haeseleer, M.; Cambron, M.; Vanopdenbosch, L.; De Keyser, J. Vascular Aspects of Multiple Sclerosis. Lancet Neurol. 2011, 10, 657–666. [Google Scholar] [CrossRef]
- Mirmosayyeb, O.; Barzegar, M.; Nehzat, N.; Shaygannejad, V.; Sahraian, M.A.; Ghajarzadeh, M. The Prevalence of Migraine in Multiple Sclerosis (MS): A Systematic Review and Meta-Analysis. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2020, 79, 33–38. [Google Scholar] [CrossRef]
- D’haeseleer, M.; Hostenbach, S.; Peeters, I.; Sankari, S.E.; Nagels, G.; De Keyser, J.; D’hooghe, M.B. Cerebral Hypoperfusion: A New Pathophysiologic Concept in Multiple Sclerosis? J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2015, 35, 1406–1410. [Google Scholar] [CrossRef]
- Senzaki, K.; Okada, Y.; Ochi, H.; Ochi, M.; Takei, S.I.; Miura, S.; Igase, M.; Ohyagi, Y. Vascular Endothelial Dysfunction Associated with Severity in Multiple Sclerosis. Mult. Scler. Relat. Disord. 2021, 54, 103135. [Google Scholar] [CrossRef]
- Lattanzi, S.; Acciarri, M.C.; Danni, M.; Taffi, R.; Cerqua, R.; Rocchi, C.; Silvestrini, M. Cerebral Hemodynamics in Patients with Multiple Sclerosis. Mult. Scler. Relat. Disord. 2020, 44, 102309. [Google Scholar] [CrossRef]
- Orian, J.M.; D’Souza, C.S.; Kocovski, P.; Krippner, G.; Hale, M.W.; Wang, X.; Peter, K. Platelets in Multiple Sclerosis: Early and Central Mediators of Inflammation and Neurodegeneration and Attractive Targets for Molecular Imaging and Site-Directed Therapy. Front. Immunol. 2021, 12, 620963. [Google Scholar] [CrossRef] [PubMed]
- van den Pol, A.N. Neuropeptide Transmission in Brain Circuits. Neuron 2012, 76, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Drew, G.M.; Coussens, C.M.; Abraham, W.C. Effects of Endothelin-1 on Hippocampal Synaptic Plasticity. Neuroreport 1998, 9, 1827–1830. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Trepanier, C.H.; Li, H.; Beazely, M.A.; Lerner, E.A.; Jackson, M.F.; MacDonald, J.F. Vasoactive Intestinal Peptide Acts via Multiple Signal Pathways to Regulate Hippocampal NMDA Receptors and Synaptic Transmission. Hippocampus 2009, 19, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Q.-Y.; Lee, J.H.; Li, X.-H.; Yu, S.; Zhuo, M. Cortical Potentiation Induced by Calcitonin Gene-Related Peptide (CGRP) in the Insular Cortex of Adult Mice. Mol. Brain 2020, 13, 36. [Google Scholar] [CrossRef]
- Gressens, P.; Hill, J.M.; Gozes, I.; Fridkin, M.; Brenneman, D.E. Growth Factor Function of Vasoactive Intestinal Peptide in Whole Cultured Mouse Embryos. Nature 1993, 362, 155–158. [Google Scholar] [CrossRef]
- Gressens, P.; Paindaveine, B.; Hill, J.M.; Brenneman, D.E.; Evrard, P. Growth Factor Properties of VIP during Early Brain Development.: Whole Embryo Culture and in Vivo Studies. Ann. N. Y. Acad. Sci. 1997, 814, 152–160. [Google Scholar] [CrossRef]
- Deng, G.; Jin, L. The Effects of Vasoactive Intestinal Peptide in Neurodegenerative Disorders. Neurol. Res. 2017, 39, 65–72. [Google Scholar] [CrossRef]
- Solés-Tarrés, I.; Cabezas-Llobet, N.; Vaudry, D.; Xifró, X. Protective Effects of Pituitary Adenylate Cyclase-Activating Polypeptide and Vasoactive Intestinal Peptide Against Cognitive Decline in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 221. [Google Scholar] [CrossRef]
- Van, C.; Condro, M.C.; Ko, H.H.; Hoang, A.Q.; Zhu, R.; Lov, K.; Ricaflanca, P.T.; Diep, A.L.; Nguyen, N.N.M.; Lipshutz, G.S.; et al. Targeted Deletion of PAC1 Receptors in Retinal Neurons Enhances Neuron Loss and Axonopathy in a Model of Multiple Sclerosis and Optic Neuritis. Neurobiol. Dis. 2021, 160, 105524. [Google Scholar] [CrossRef]
- Abad, C.; Jayaram, B.; Becquet, L.; Wang, Y.; O’Dorisio, M.S.; Waschek, J.A.; Tan, Y.-V. VPAC1 Receptor (Vipr1)-Deficient Mice Exhibit Ameliorated Experimental Autoimmune Encephalomyelitis, with Specific Deficits in the Effector Stage. J. Neuroinflamm. 2016, 13, 169. [Google Scholar] [CrossRef]
- Masaki, T.; Sawamura, T. Endothelin and Endothelial Dysfunction. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2006, 82, 17–24. [Google Scholar] [CrossRef]
- Masaki, T. Historical Review: Endothelin. Trends Pharmacol. Sci. 2004, 25, 219–224. [Google Scholar] [CrossRef]
- Marola, O.J.; Syc-Mazurek, S.B.; Howell, G.R.; Libby, R.T. Endothelin 1-Induced Retinal Ganglion Cell Death Is Largely Mediated by JUN Activation. Cell Death Dis. 2020, 11, 811. [Google Scholar] [CrossRef]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-Derived Endothelin-1 Inhibits Remyelination through Notch Activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; McEllin, B.; Morton, P.D.; Raymond, M.; Dupree, J.; Gallo, V. Endothelin-B Receptor Activation in Astrocytes Regulates the Rate of Oligodendrocyte Regeneration during Remyelination. Cell Rep. 2015, 13, 2090–2097. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Peng, L.; Yan, B.; Dong, Q.; Seng, Z.; Wang, W.; Lv, J.; He, X. Bosentan Reduces Neuronal Apoptosis Following Spinal Cord Ischemic Reperfusion Injury. Spinal Cord 2014, 52, 181–185. [Google Scholar] [CrossRef] [PubMed]
- He, J.-T.; Li, H.; Yang, L.; Cheng, K.-L. Involvement of Endothelin-1, H2S and Nrf2 in Beneficial Effects of Remote Ischemic Preconditioning in Global Cerebral Ischemia-Induced Vascular Dementia in Mice. Cell. Mol. Neurobiol. 2019, 39, 671–686. [Google Scholar] [CrossRef]
- Jin, Y.-H.; Kang, B.; Kang, H.S.; Koh, C.-S.; Kim, B.S. Endothelin-1 Contributes to the Development of Virus-Induced Demyelinating Disease. J. Neuroinflamm. 2020, 17, 307. [Google Scholar] [CrossRef]
- Guo, Y.; Chung, S.K.; Siu, C.-W.; Kwan, S.-C.; Ho, P.W.-L.; Yeung, P.K.-K.; Chan, K.-H. Endothelin-1 Overexpression Exacerbate Experimental Allergic Encephalomyelitis. J. Neuroimmunol. 2014, 276, 64–70. [Google Scholar] [CrossRef]
- Castellazzi, M.; Lamberti, G.; Resi, M.V.; Baldi, E.; Caniatti, L.M.; Galante, G.; Perri, P.; Pugliatti, M. Increased Levels of Endothelin-1 in Cerebrospinal Fluid Are a Marker of Poor Visual Recovery after Optic Neuritis in Multiple Sclerosis Patients. Dis. Markers 2019, 2019, 9320791. [Google Scholar] [CrossRef] [PubMed]
- Monti, L.; Arrigucci, U.; Rossi, A. Insights into Endothelin-3 and Multiple Sclerosis. Biomol. Concepts 2020, 11, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rey, E.; Fernandez-Martin, A.; Chorny, A.; Martin, J.; Pozo, D.; Ganea, D.; Delgado, M. Therapeutic Effect of Vasoactive Intestinal Peptide on Experimental Autoimmune Encephalomyelitis. Am. J. Pathol. 2006, 168, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Ito, A.; Kawanokuchi, J.; Jin, S.; Mizuno, T.; Ojika, K.; Ueda, R.; Suzumura, A. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Ameliorates Experimental Autoimmune Encephalomyelitis by Suppressing the Functions of Antigen Presenting Cells. Mult. Scler. Houndmills Basingstoke Engl. 2004, 10, 651–659. [Google Scholar] [CrossRef]
- Rossetti, I.; Zambusi, L.; Finardi, A.; Bodini, A.; Provini, L.; Furlan, R.; Morara, S. Calcitonin Gene-Related Peptide Decreases IL-1beta, IL-6 as Well as Ym1, Arg1, CD163 Expression in a Brain Tissue Context-Dependent Manner While Ameliorating Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol. 2018, 323, 94–104. [Google Scholar] [CrossRef]
- Russell, F.A.; King, R.; Smillie, S.J.; Kodji, X.; Brain, S.D. Calcitonin Gene-Related Peptide: Physiology and Pathophysiology. Physiol. Rev. 2014, 94, 1099–1142. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, N.; Chen, L.; Zhao, X.; Li, M.-R. Independent Roles of CGRP in Cardioprotection and Hemodynamic Regulation in Ischemic Postconditioning. Eur. J. Pharmacol. 2018, 828, 18–25. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Q.; Cai, H.; Xu, C.; Liu, G.; Li, Z. Calcitonin Gene-Related Peptide Prevents Blood–Brain Barrier Injury and Brain Edema Induced by Focal Cerebral Ischemia Reperfusion. Regul. Pept. 2011, 171, 19–25. [Google Scholar] [CrossRef]
- Zhai, L.; Sakurai, T.; Kamiyoshi, A.; Ichikawa-Shindo, Y.; Kawate, H.; Tanaka, M.; Xian, X.; Hirabayashi, K.; Dai, K.; Cui, N.; et al. Endogenous Calcitonin Gene-Related Peptide Suppresses Ischemic Brain Injuries and Progression of Cognitive Decline. J. Hypertens. 2018, 36, 876–891. [Google Scholar] [CrossRef]
- Hay, D.L.; Garelja, M.L.; Poyner, D.R.; Walker, C.S. Update on the Pharmacology of Calcitonin/CGRP Family of Peptides: IUPHAR Review 25. Br. J. Pharmacol. 2018, 175, 3–17. [Google Scholar] [CrossRef]
- Gingell, J.J.; Hendrikse, E.R.; Hay, D.L. New Insights into the Regulation of CGRP-Family Receptors. Trends Pharmacol. Sci. 2019, 40, 71–83. [Google Scholar] [CrossRef]
- Borkum, J.M. CGRP and Brain Functioning: Cautions for Migraine Treatment. Headache 2019, 59, 1339–1357. [Google Scholar] [CrossRef]
- Piehl, F.; Arvidsson, U.; Johnson, H.; Cullheim, S.; Dagerlind, A.; Ulfhake, B.; Cao, Y.; Elde, R.; Pettersson, R.F.; Terenius, L. GAP-43, AFGF, CCK and Alpha- and Beta-CGRP in Rat Spinal Motoneurons Subjected to Axotomy and/or Dorsal Root Severance. Eur. J. Neurosci. 1993, 5, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Giardino, L.; Giuliani, A.; Fernandez, M.; Calzà, L. Spinal Motoneurone Distress during Experimental Allergic Encephalomyelitis. Neuropathol. Appl. Neurobiol. 2004, 30, 522–531. [Google Scholar] [CrossRef]
- Mathé, A.A.; Ågren, H.; Lindström, L.; Theodorsson, E. Increased Concentration of Calcitonin Gene-Related Peptide in Cerebrospinal Fluid of Depressed Patients. A Possible Trait Marker of Major Depressive Disorder. Neurosci. Lett. 1994, 182, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Mathé, A.A.; Hertel, P.; Nomikos, G.G.; Gruber, S.; Mathé, J.M.; Svensson, T.H. The Psychotomimetic Drugs D-Amphetamine and Phencyclidine Release Calcitonin Gene-Related Peptide in the Limbic Forebrain of the Rat. J. Neurosci. Res. 1996, 46, 316–323. [Google Scholar] [CrossRef]
- Siegert, R.J.; Abernethy, D.A. Depression in Multiple Sclerosis: A Review. J. Neurol. Neurosurg. Psychiatry 2005, 76, 469–475. [Google Scholar] [CrossRef]
- Warfvinge, K.; Edvinsson, L.; Pickering, D.S.; Sheykhzade, M. The Presence of Calcitonin Gene-Related Peptide and Its Receptors in Rat, Pig and Human Brain: Species Differences in Calcitonin Gene-Related Peptide Pharmacology. Pharmacology 2019, 104, 332–341. [Google Scholar] [CrossRef]
- Kaiser, E.A.; Rea, B.J.; Kuburas, A.; Kovacevich, B.R.; Garcia-, L.F.; Recober, A.; Russo, A.F.; City, I.; States, U.; City, I.; et al. Negative Regulation of TLR Responses by the Neuropeptide CGRP Is Mediated by the Transcriptional Repressor ICER. Sci. Rep. 2017, 323, 607–615. [Google Scholar] [CrossRef]
- Kee, Z.; Kodji, X.; Brain, S.D. The Role of Calcitonin Gene Related Peptide (CGRP) in Neurogenic Vasodilation and Its Cardioprotective Effects. Front. Physiol. 2018, 9, 1249. [Google Scholar] [CrossRef]
- Pedreño, M.; Morell, M.; Robledo, G.; Souza-Moreira, L.; Forte-Lago, I.; Caro, M.; O’Valle, F.; Ganea, D.; Gonzalez-Rey, E. Adrenomedullin Protects from Experimental Autoimmune Encephalomyelitis at Multiple Levels. Brain. Behav. Immun. 2014, 37, 152–163. [Google Scholar] [CrossRef]
- Rodríguez-Lorenzo, S.; Ferreira Francisco, D.M.; Vos, R.; van Het Hof, B.; Rijnsburger, M.; Schroten, H.; Ishikawa, H.; Beaino, W.; Bruggmann, R.; Kooij, G.; et al. Altered Secretory and Neuroprotective Function of the Choroid Plexus in Progressive Multiple Sclerosis. Acta Neuropathol. Commun. 2020, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.L.; Chen, S.; Lutz, T.A.; Parkes, D.G.; Roth, J.D. Amylin: Pharmacology, Physiology, and Clinical Potential. Pharmacol. Rev. 2015, 67, 564–600. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Zhu, H.; Wang, X.; Gower, A.C.; Wallack, M.; Blusztajn, J.K.; Kowall, N.; Qiu, W.Q. Amylin Treatment Reduces Neuroinflammation and Ameliorates Abnormal Patterns of Gene Expression in the Cerebral Cortex of an Alzheimer’s Disease Mouse Model. J. Alzheimers Dis. 2017, 56, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Grizzanti, J.; Corrigan, R.; Casadesus, G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J. Alzheimers Dis. JAD 2018, 66, 11–23. [Google Scholar] [CrossRef]
- Walker, C.S.; Conner, A.C.; Poyner, D.R.; Hay, D.L. Regulation of Signal Transduction by Calcitonin Gene-Related Peptide Receptors. Trends Pharmacol. Sci. 2010, 31, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Kurnellas, M.P.; Adams, C.M.; Sobel, R.A.; Steinman, L.; Rothbard, J.B. Amyloid Fibrils Composed of Hexameric Peptides Attenuate Neuroinflammation. Sci. Transl. Med. 2013, 5, 179ra42. [Google Scholar] [CrossRef]
- Steinman, L.; Rothbard, J.B.; Kurnellas, M.P. Janus Faces of Amyloid Proteins in Neuroinflammation. J. Clin. Immunol. 2014, 34 (Suppl 1), S61–S63. [Google Scholar] [CrossRef]
- Pietroboni, A.M.; Colombi, A.; Carandini, T.; Scarpini, E.; Galimberti, D.; Bozzali, M. The Role of Amyloid-ß in White Matter Damage: Possible Common Pathogenetic Mechanisms in Neurodegenerative and Demyelinating Diseases. J. Alzheimers Dis. 2020, 78, 13–22. [Google Scholar] [CrossRef]
- Pietroboni, A.M.; Caprioli, M.; Carandini, T.; Scarioni, M.; Ghezzi, L.; Arighi, A.; Cioffi, S.; Cinnante, C.; Fenoglio, C.; Oldoni, E.; et al. CSF β-Amyloid Predicts Prognosis in Patients with Multiple Sclerosis. Mult. Scler. Houndmills Basingstoke Engl. 2019, 25, 1223–1231. [Google Scholar] [CrossRef]
- Bozkurt, B.; Nair, A.P.; Misra, A.; Scott, C.Z.; Mahar, J.H.; Fedson, S. Neprilysin Inhibitors in Heart Failure. JACC Basic Transl. Sci. 2022, 8, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, D.; Adornetto, A.; Rombolà, L.; Naturale, M.D.; Morrone, L.A.; Bagetta, G.; Tonin, P.; Corasaniti, M.T. New Trends in Migraine Pharmacology: Targeting Calcitonin Gene–Related Peptide (CGRP) With Monoclonal Antibodies. Front. Pharmacol. 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Hersh, L.B.; Rodgers, D.W. Neprilysin and Amyloid Beta Peptide Degradation. Curr. Alzheimer Res. 2008, 5, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Chow, K.M.; Shah, R.; Rhodes, C.J.; Hersh, L.B. Degradation of Islet Amyloid Polypeptide by Neprilysin. Diabetologia 2012, 55, 2989–2998. [Google Scholar] [CrossRef]
- Eckman, E.A.; Reed, D.K.; Eckman, C.B. Degradation of the Alzheimer’s Amyloid Beta Peptide by Endothelin-Converting Enzyme. J. Biol. Chem. 2001, 276, 24540–24548. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Guarnera, E.; Berezovsky, I.N. Insulin-Degrading Enzyme in the Fight against Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 49–58. [Google Scholar] [CrossRef]
- Sousa, L.; Guarda, M.; Meneses, M.J.; Macedo, M.P.; Vicente Miranda, H. Insulin-Degrading Enzyme: An Ally against Metabolic and Neurodegenerative Diseases. J. Pathol. 2021, 255, 346–361. [Google Scholar] [CrossRef]
- Graykowski, D.; Kasparian, K.; Caniglia, J.; Gritsaeva, Y.; Cudaback, E. Neuroinflammation Drives APOE Genotype-Dependent Differential Expression of Neprilysin. J. Neuroimmunol. 2020, 346, 577315. [Google Scholar] [CrossRef]
- Zagon, I.S.; McLaughlin, P.J. Endogenous Opioids in the Etiology and Treatment of Multiple Sclerosis; Exon Publications: Brisbane City, Australia, 2017; ISBN 978-0-9944381-3-3. [Google Scholar]
- Ban, M.; McCauley, J.L.; Zuvich, R.; Baker, A.; Bergamaschi, L.; Cox, M.; Kemppinen, A.; D’Alfonso, S.; Guerini, F.R.; Lechner-Scott, J.; et al. A Non-Synonymous SNP within Membrane Metalloendopeptidase-like 1 (MMEL1) Is Associated with Multiple Sclerosis. Genes Immun. 2010, 11, 660–664. [Google Scholar] [CrossRef]
- Bernstein, H.-G.; Keilhoff, G.; Dobrowolny, H.; Steiner, J. The Many Facets of CD26/Dipeptidyl Peptidase 4 and Its Inhibitors in Disorders of the CNS—A Critical Overview. Rev. Neurosci. 2022, 34. [Google Scholar] [CrossRef]
- Tejera-Alhambra, M.; Casrouge, A.; de Andrés, C.; Ramos-Medina, R.; Alonso, B.; Vega, J.; Albert, M.L.; Sánchez-Ramón, S. Low DPP4 Expression and Activity in Multiple Sclerosis. Clin. Immunol. Orlando Fla 2014, 150, 170–183. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papiri, G.; D’Andreamatteo, G.; Cacchiò, G.; Alia, S.; Silvestrini, M.; Paci, C.; Luzzi, S.; Vignini, A. Multiple Sclerosis: Inflammatory and Neuroglial Aspects. Curr. Issues Mol. Biol. 2023, 45, 1443-1470. https://doi.org/10.3390/cimb45020094
Papiri G, D’Andreamatteo G, Cacchiò G, Alia S, Silvestrini M, Paci C, Luzzi S, Vignini A. Multiple Sclerosis: Inflammatory and Neuroglial Aspects. Current Issues in Molecular Biology. 2023; 45(2):1443-1470. https://doi.org/10.3390/cimb45020094
Chicago/Turabian StylePapiri, Giulio, Giordano D’Andreamatteo, Gabriella Cacchiò, Sonila Alia, Mauro Silvestrini, Cristina Paci, Simona Luzzi, and Arianna Vignini. 2023. "Multiple Sclerosis: Inflammatory and Neuroglial Aspects" Current Issues in Molecular Biology 45, no. 2: 1443-1470. https://doi.org/10.3390/cimb45020094
APA StylePapiri, G., D’Andreamatteo, G., Cacchiò, G., Alia, S., Silvestrini, M., Paci, C., Luzzi, S., & Vignini, A. (2023). Multiple Sclerosis: Inflammatory and Neuroglial Aspects. Current Issues in Molecular Biology, 45(2), 1443-1470. https://doi.org/10.3390/cimb45020094