
The Potential Inhibitory Role of Acetyl-L-Carnitine on Proliferation, Migration, and Gene Expression in HepG2 and HT29 Human Adenocarcinoma Cell Lines

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells Culture and Reagents
2.2. Cell Viability Assay
2.3. Migration Assay
2.4. Detection of Apoptotic DNA
2.5. Fluorescent Microscopy for Evaluating Apoptosis
2.6. RNA Extraction and Reverse Transcription-Real-Time Polymerase Chain Reaction
2.7. Statistical Analysis
3. Results
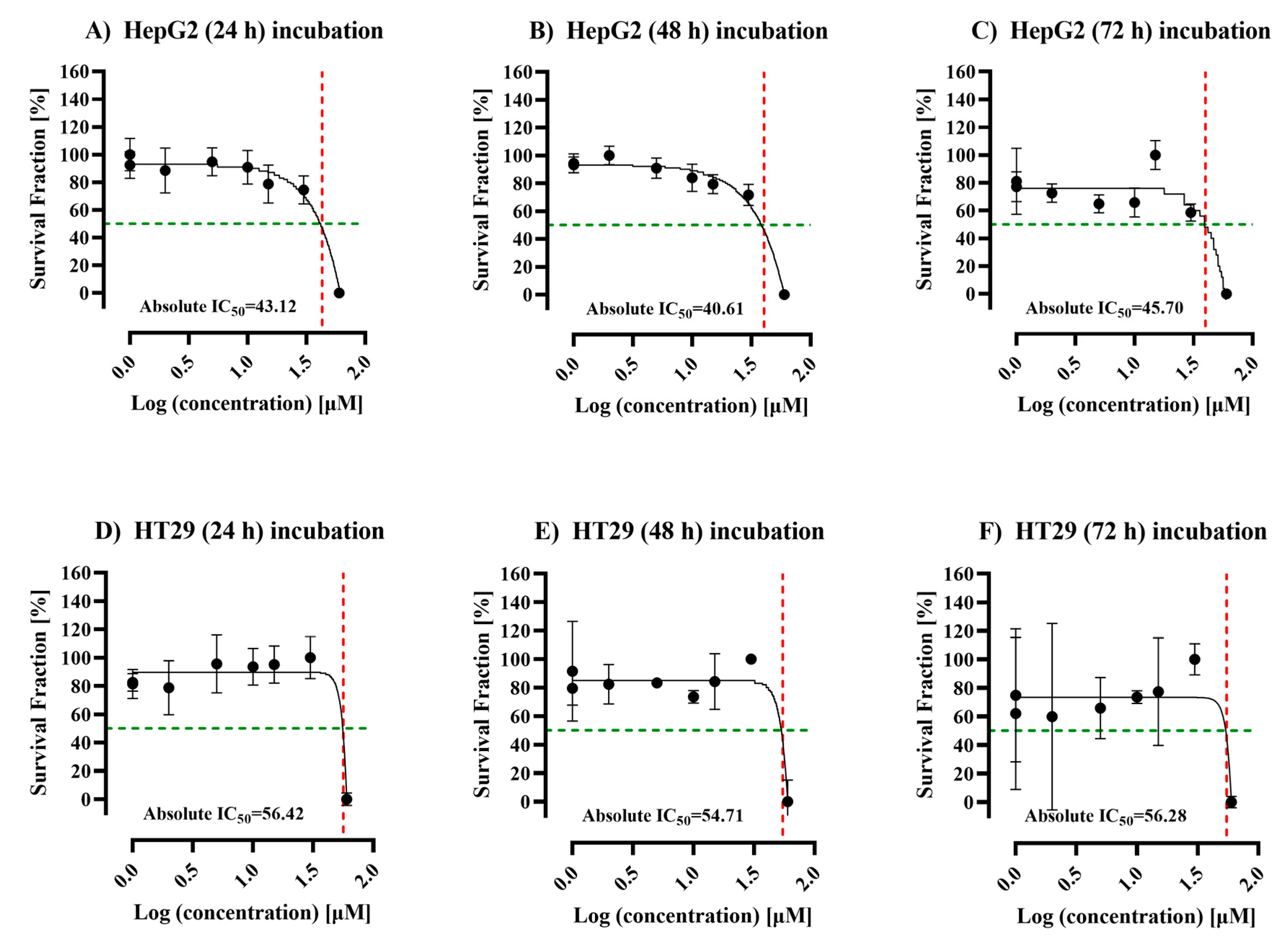
3.1. Efficacy of ALC on the Viability of HepG2 and HT29 Cell Lines
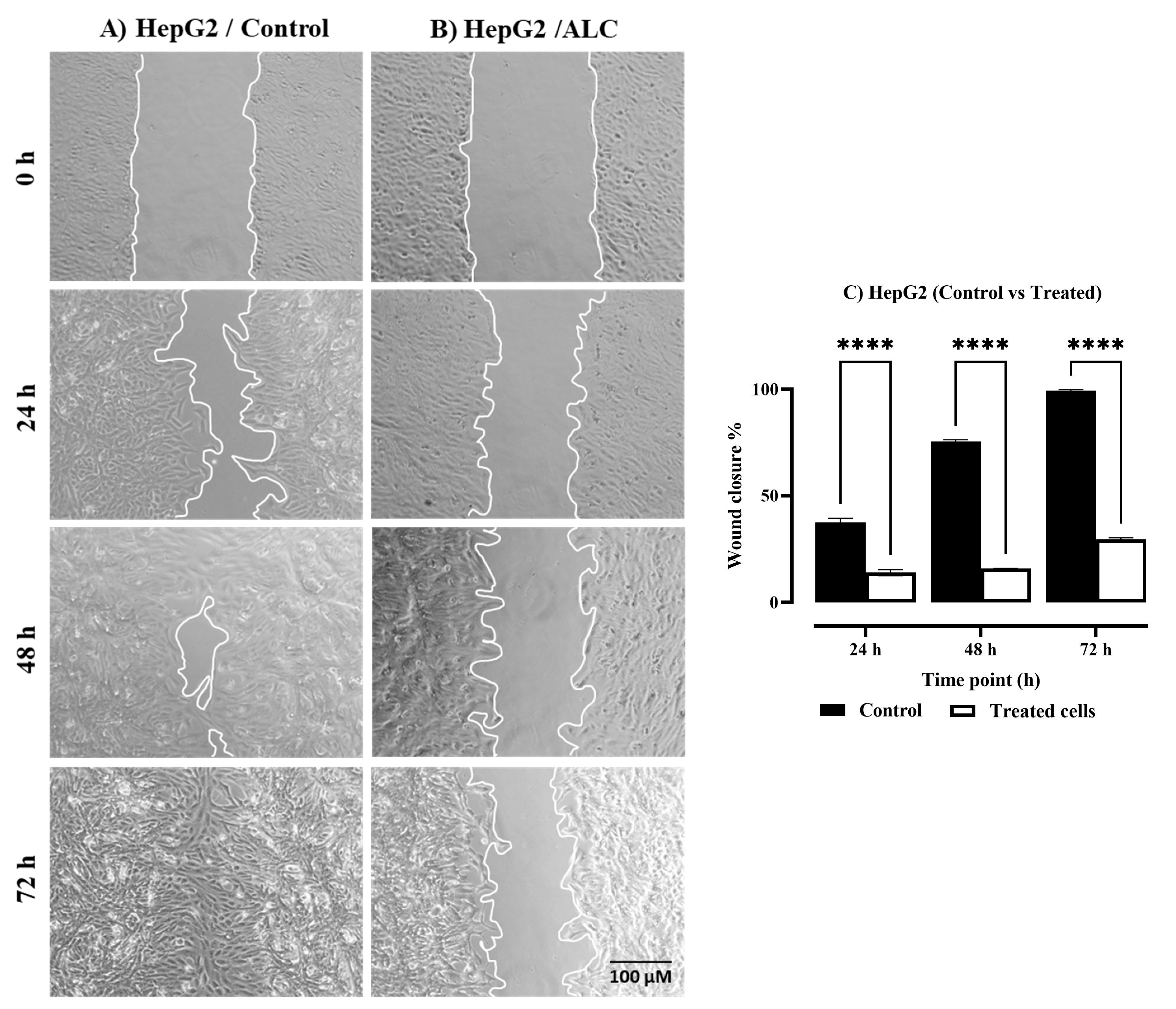
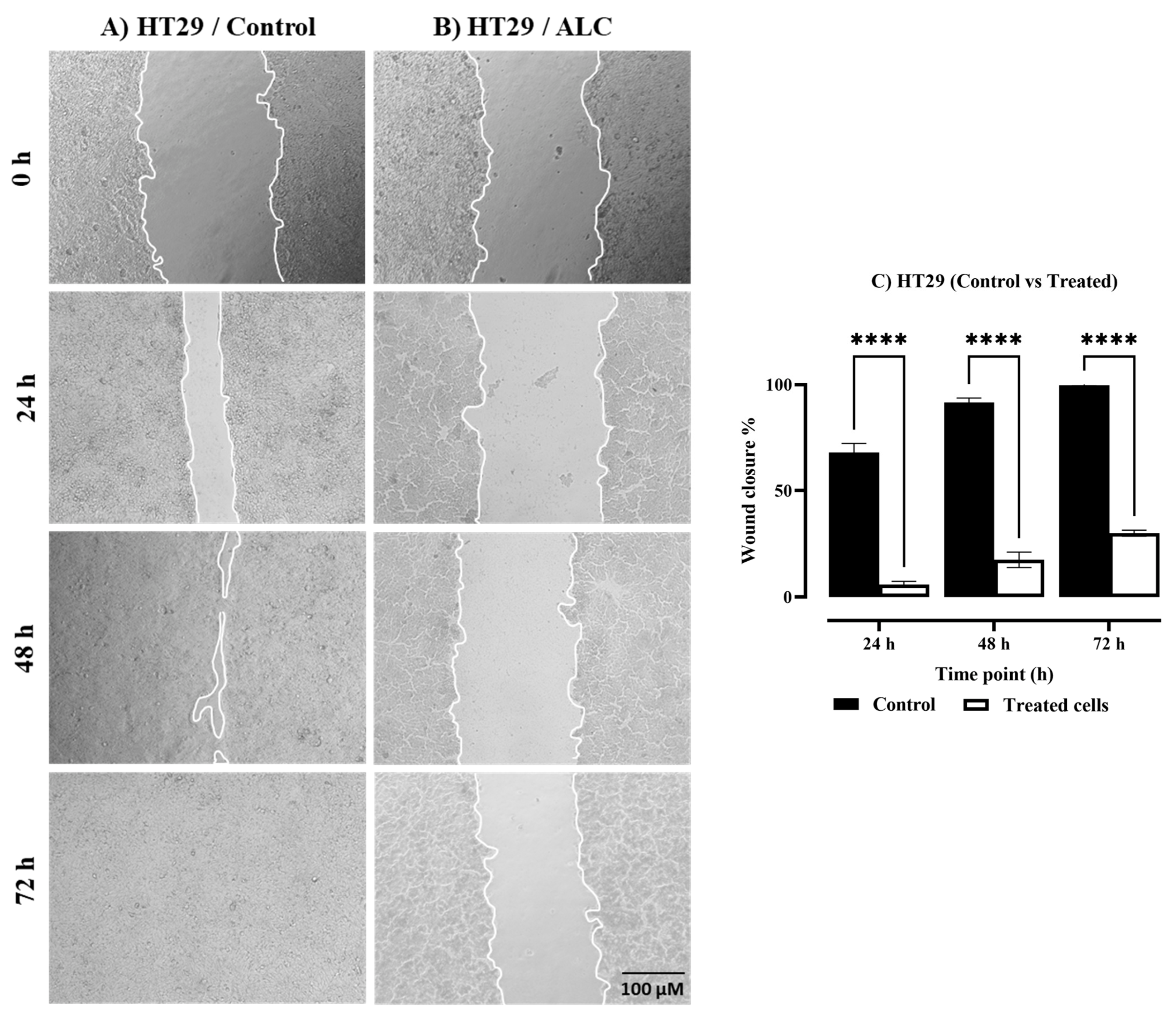
3.2. ALC Reduced Migration in HepG2 and HT29 Cell Lines
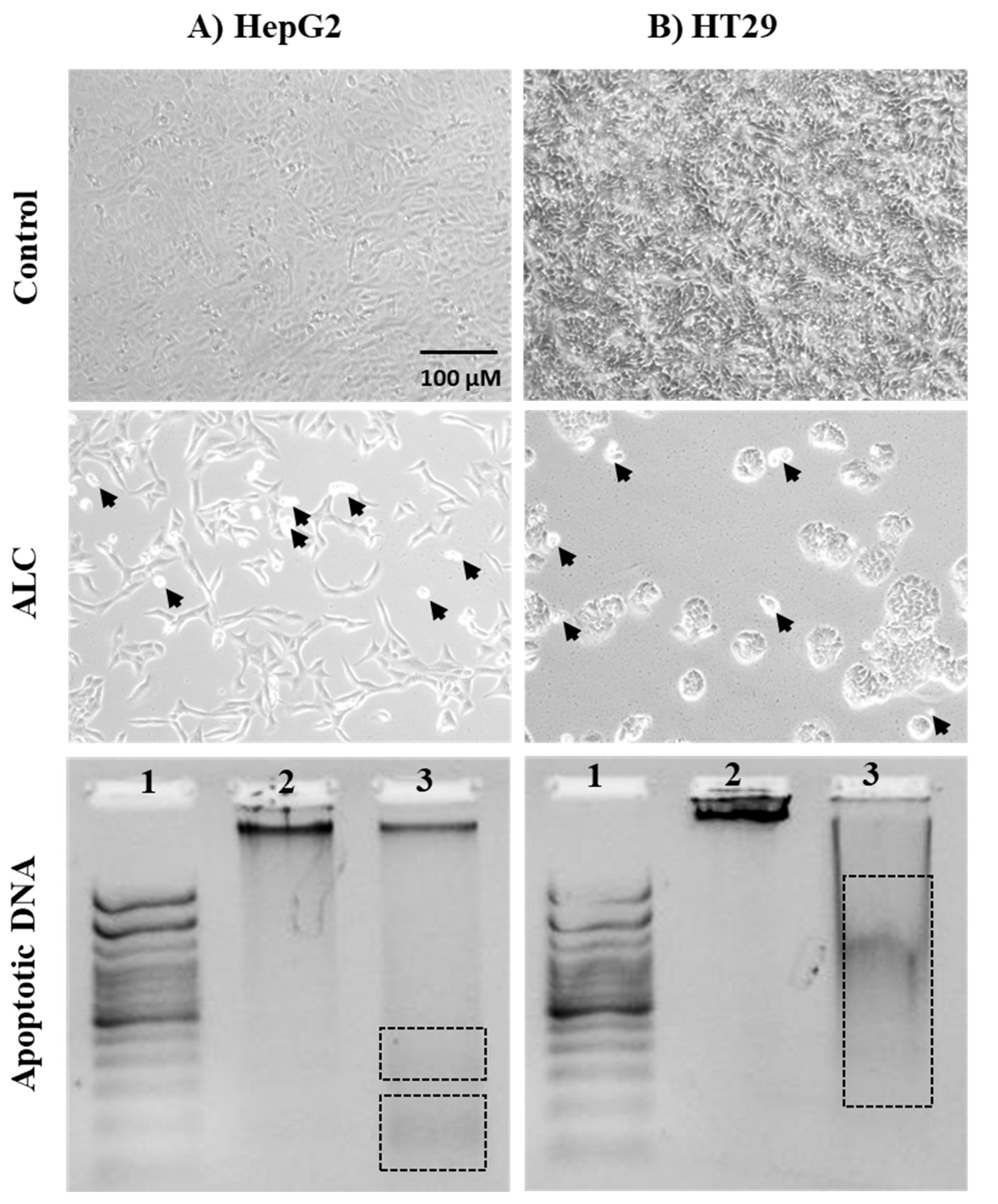
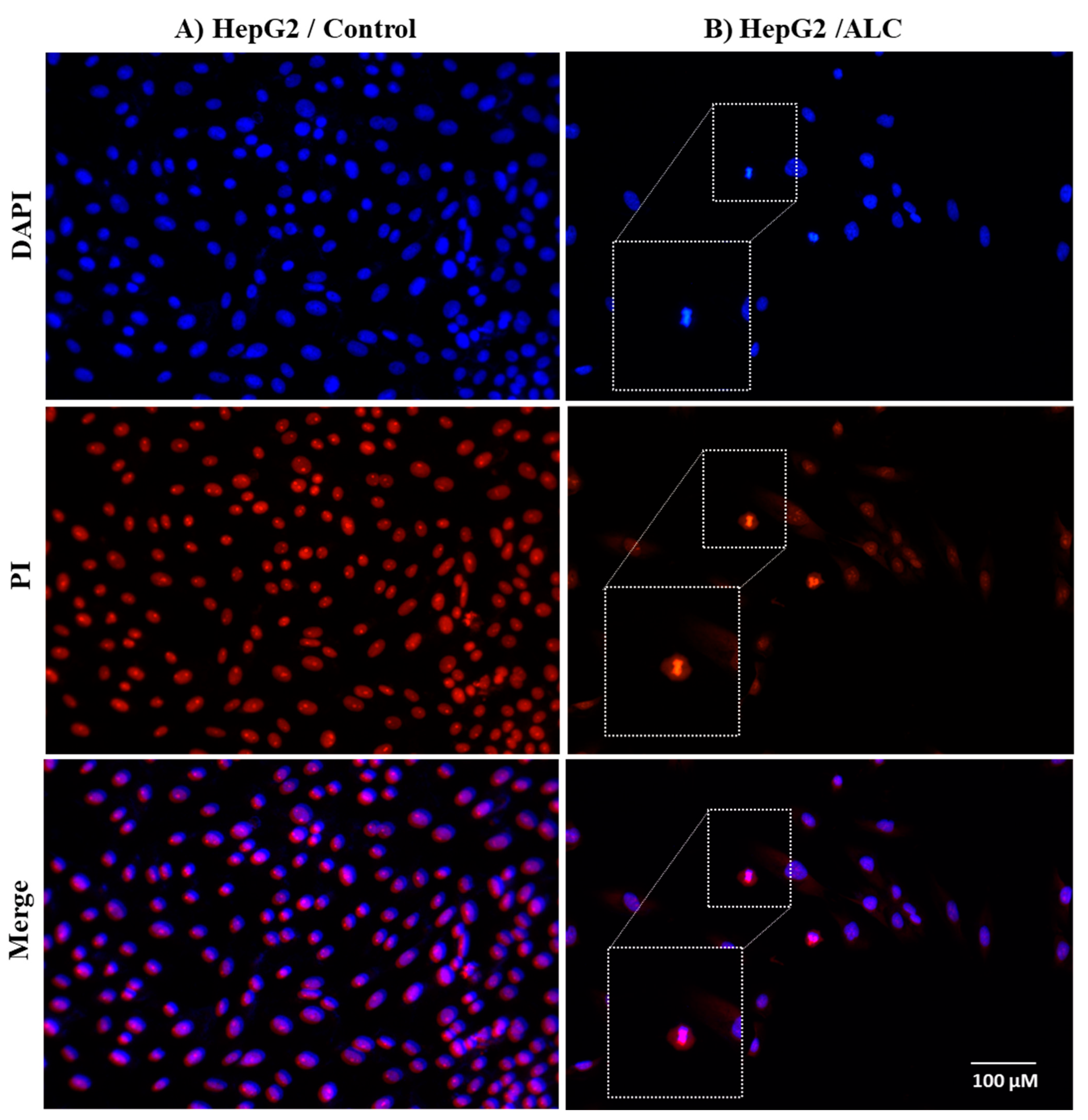
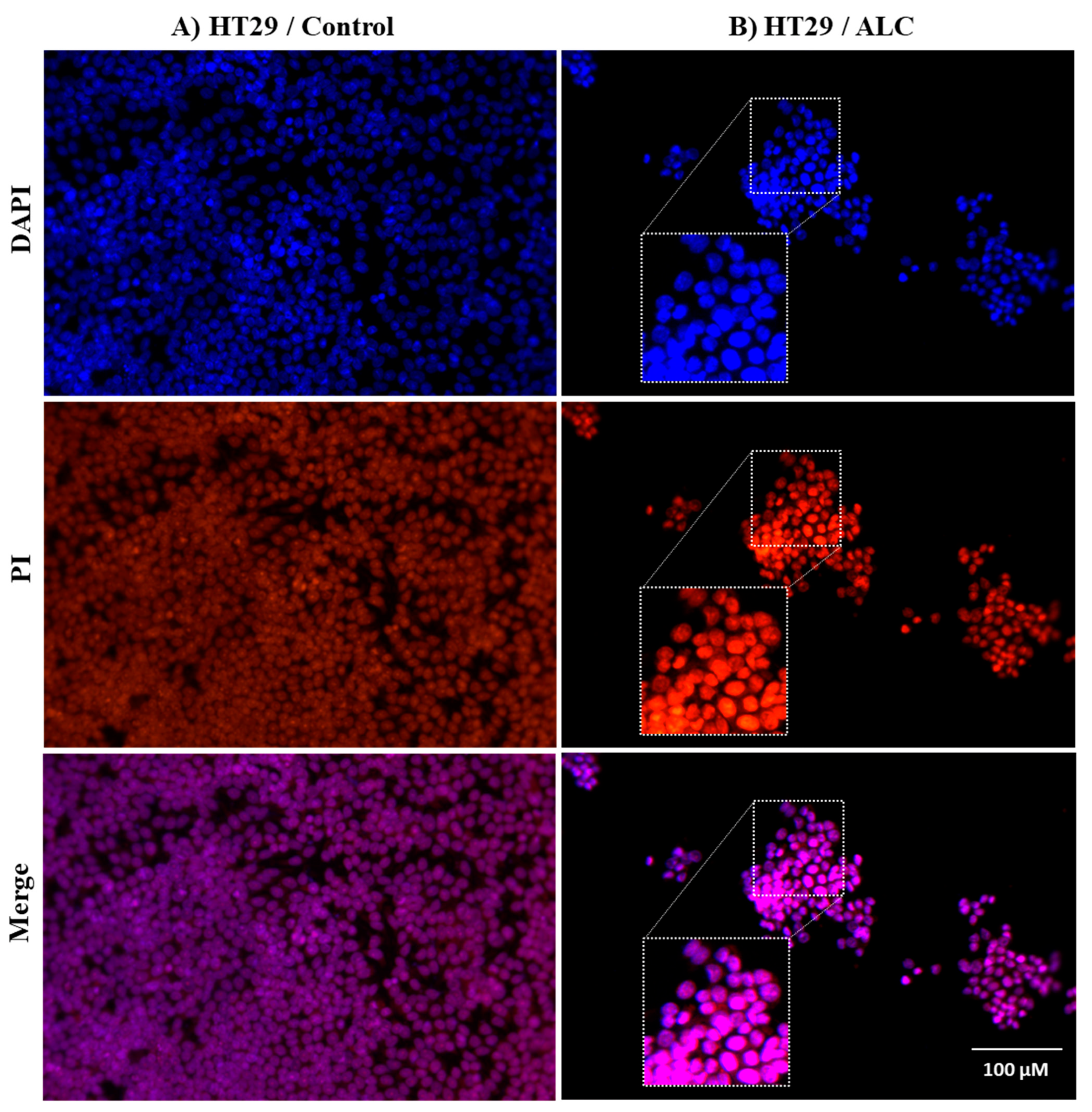
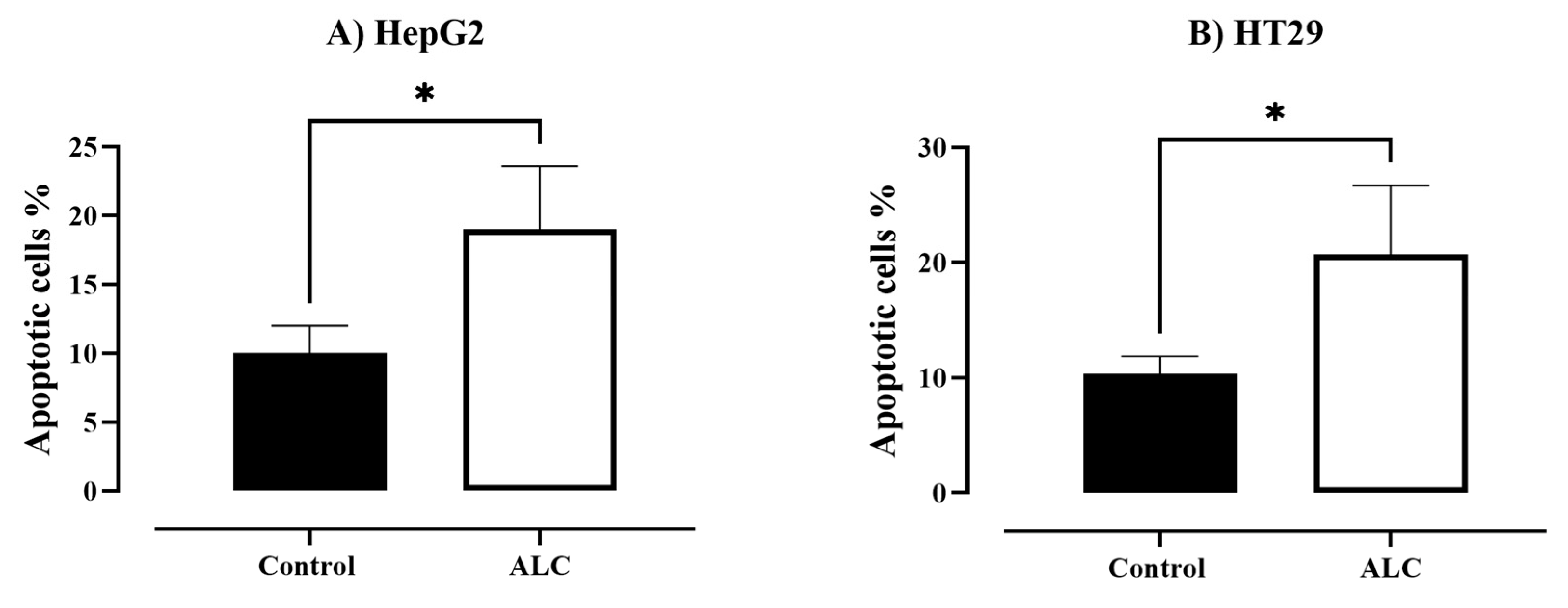
3.3. ALC Influences Nucleus Morphology in HepG2 and HT29 Cell Lines and Induced DNA Fragmentation
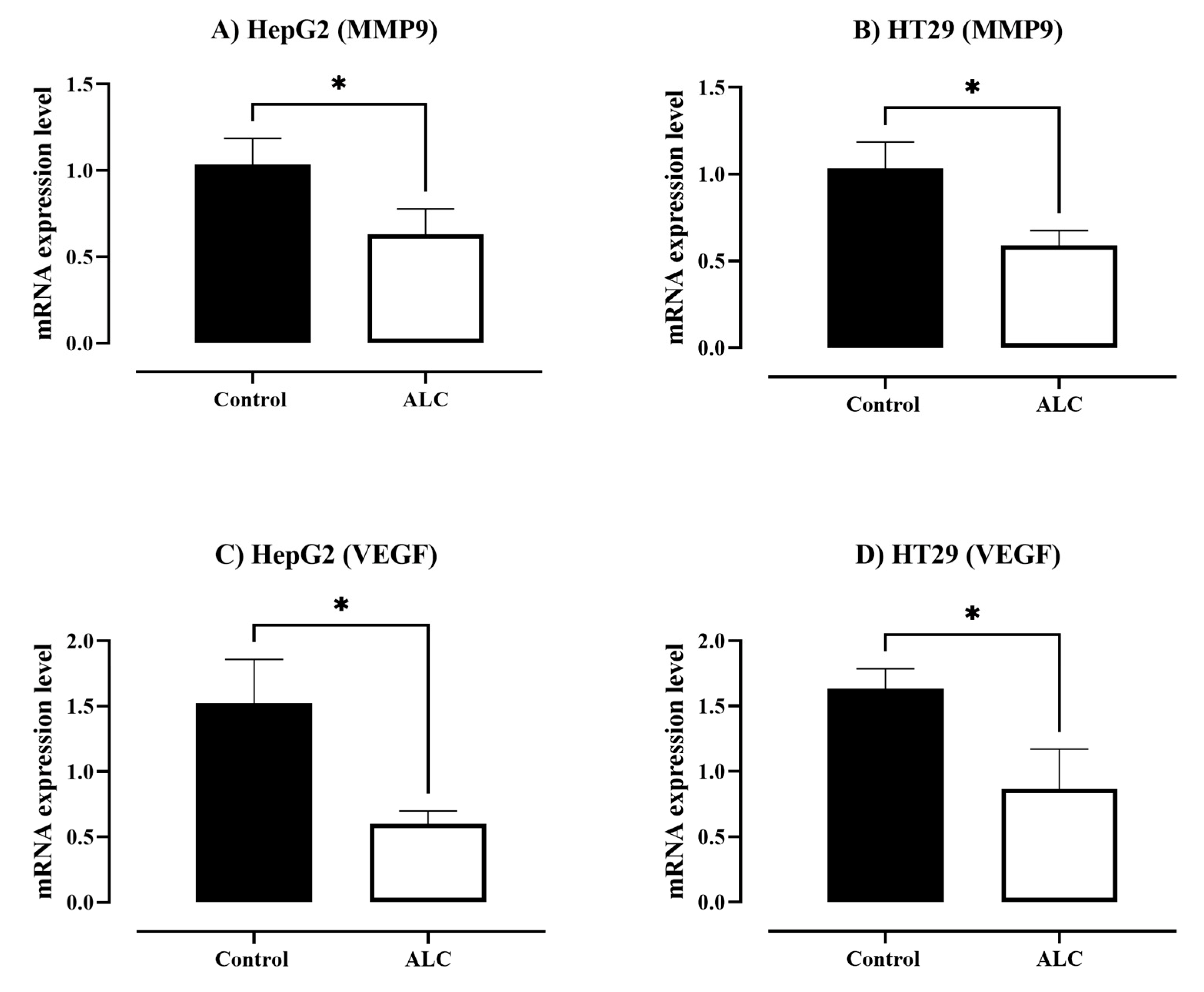
3.4. ALC Downregulates the Expression Level of Matrix Metallopeptidase 9 (MMP9) and Vascular Endothelial Growth Factor (VEGF) in HepG2 and HT29 Cell Lines
4. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herszenyi, L.; Tulassay, Z. Epidemiology of gastrointestinal and liver tumors. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 249–258. [Google Scholar]
- Pinheiro, P.S.; Callahan, K.E.; Jones, P.D.; Morris, C.; Ransdell, J.M.; Kwon, D.; Brown, C.P.; Kobetz, E.N. Liver cancer: A leading cause of cancer death in the United States and the role of the 1945–1965 birth cohort by ethnicity. JHEP Rep. 2019, 1, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Alqahtani, A.; Khan, Z.; Alloghbi, A.; Said Ahmed, T.S.; Ashraf, M.; Hammouda, D.M. Hepatocellular carcinoma: Molecular mechanisms and targeted therapies. Medicina 2019, 55, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; Maclntyre, M.; Allen, C.; Hansen, G.; Woodbrook, R.; Wolfe, C. The global burden of cancer 2013 global burden of disease cancer collaboration. JAMA Oncol. 2015, 1, 505–527. [Google Scholar]
- Liu, P.; Xie, S.-H.; Hu, S.; Cheng, X.; Gao, T.; Zhang, C.; Song, Z. Age-specific sex difference in the incidence of hepatocellular carcinoma in the United States. Oncotarget 2017, 8, 68131. [Google Scholar] [CrossRef] [Green Version]
- Carr, B.I.; Nagalla, S.; Geva, R. Medical therapy of HCC. In Hepatocellular Carcinoma; Springer: Berlin/Heidelberg, Germany, 2016; pp. 489–512. [Google Scholar]
- McGlynn, K.A.; London, W.T. The global epidemiology of hepatocellular carcinoma: Present and future. Clin. Liver Dis. 2011, 15, 223–243. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.W.; Kroker-Lobos, M.F.; Lazo, M.; Rivera-Andrade, A.; Egner, P.A.; Wedemeyer, H.; Torres, O.; Freedman, N.D.; McGlynn, K.A.; Guallar, E. Aflatoxin and viral hepatitis exposures in Guatemala: Molecular biomarkers reveal a unique profile of risk factors in a region of high liver cancer incidence. PLoS ONE 2017, 12, e0189255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA A Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Wu, K. Cancer epidemiology and prevention. In Cancers of the Colon and Rectum, 3rd ed.; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Botteri, E.; Iodice, S.; Bagnardi, V.; Raimondi, S.; Lowenfels, A.B.; Maisonneuve, P. Smoking and colorectal cancer: A meta-analysis. JAMA 2008, 300, 2765–2778. [Google Scholar] [CrossRef]
- Giovannucci, E. Modifiable risk factors for colon cancer. Gastroenterol. Clin. 2002, 31, 925–943. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [Green Version]
- Wattenberg, L.W. Inhibition of carcinogenesis by minor dietary constituents. Cancer Res. 1992, 52, 2085s–2091s. [Google Scholar] [PubMed]
- Kostova, I. Synthetic and natural coumarins as cytotoxic agents. Curr. Med. Chem.-Anti-Cancer Agents 2005, 5, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. Part C Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- İzgüt-Uysal, V.N.; Bülbül, M.; Tan, R.; Üstünel, İ.; Ağar, A.; Yargiçoğlu, P. Effect of chronic stress and L-carnitine on rat stomach. J. Physiol. Sci. 2007, 57, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Gülcin, İ. Antioxidant and antiradical activities of L-carnitine. Life Sci. 2006, 78, 803–811. [Google Scholar] [CrossRef]
- Peluso, G.; Nicolai, R.; Reda, E.; Benatti, P.; Barbarisi, A.; Calvani, M. Cancer and anticancer therapy-induced modifications on metabolism mediated by carnitine system. J. Cell. Physiol. 2000, 182, 339–350. [Google Scholar] [CrossRef]
- Wang, M.; Wang, K.; Liao, X.; Hu, H.; Chen, L.; Meng, L.; Gao, W.; Li, Q. Carnitine Palmitoyltransferase system: A new target for anti-inflammatory and anticancer therapy? Front. Pharmacol. 2021, 12, 760581. [Google Scholar] [CrossRef] [PubMed]
- Radkhouy, F.; Soltanieh, S.; Solgi, S.; Ansari, M.; Abbasi, B. The effect of L-carnitine on colorectal cancer: A review on current evidence. Food Health 2018, 1, 39–47. [Google Scholar]
- Flanagan, J.L.; Simmons, P.A.; Vehige, J.; Willcox, M.D.; Garrett, Q. Role of carnitine in disease. Nutr. Metab. 2010, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.-J.; Dionne, S.; Marx, G.; Qureshi, I.; Sarma, D.; Levy, E.; Seidman, E.G. In vitro studies on the inhibition of colon cancer by butyrate and carnitine. Nutrition 2009, 25, 1193–1201. [Google Scholar] [CrossRef]
- Dionne, S.; Elimrani, I.; Roy, M.-J.; Qureshi, I.A.; Sarma, D.R.; Levy, E.; Seidman, E.G. Studies on the chemopreventive effect of carnitine on tumorigenesis in vivo, using two experimental murine models of colon cancer. Nutr. Cancer 2012, 64, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.I.; Pita, I.; Águas, R.; Tagorti, S.; Virmani, A.; Pereira, F.C.; Rego, A.C. Mechanistic perspectives on differential mitochondrial-based neuroprotective effects of several carnitine forms in Alzheimer’s disease in vitro model. Arch. Toxicol. 2021, 95, 2769–2784. [Google Scholar] [CrossRef] [PubMed]
- Mazdeh, M.; Abolfathi, P.; Sabetghadam, M.; Mohammadi, Y.; Mehrpooya, M. Clinical evidence of acetyl-l-carnitine efficacy in the treatment of acute ischemic stroke: A pilot clinical trial. Oxidative Med. Cell. Longev. 2022, 2022, 2493053. [Google Scholar] [CrossRef]
- Pettegrew, J.; Levine, J.; McClure, R. Acetyl-L-carnitine physical-chemical, metabolic, and therapeutic properties: Relevance for its mode of action in Alzheimer’s disease and geriatric depression. Mol. Psychiatry 2000, 5, 616–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeva-Andany, M.; Calvo-Castro, I.; Fernández-Fernández, C.; Donapetry-García, C. Pedre-Piñeiro A. Significance of l-carnitine for human health. IUBMB Life 2017, 69, 578–594. [Google Scholar] [CrossRef] [Green Version]
- Baci, D.; Bruno, A.; Cascini, C.; Gallazzi, M.; Mortara, L.; Sessa, F.; Pelosi, G.; Albini, A.; Noonan, D.M. Acetyl-L-Carnitine downregulates invasion (CXCR4/CXCL12, MMP-9) and angiogenesis (VEGF, CXCL8) pathways in prostate cancer cells: Rationale for prevention and interception strategies. J. Exp. Clin. Cancer Res. 2019, 38, 464. [Google Scholar] [CrossRef]
- Pan, T.; Qian, Y.; Li, T.; Zhang, Z.; He, Y.; Wang, J.; Li, L.; Hu, Y.; Lin, M. Acetyl l-carnitine protects adipose-derived stem cells against serum-starvation: Regulation on the network composed of reactive oxygen species, autophagy, apoptosis and senescence. Cytotechnology 2022, 74, 105–121. [Google Scholar] [CrossRef]
- Wang, S.; Xu, J.; Zheng, J.; Zhang, X.; Shao, J.; Zhao, L.; Hao, J. Anti-inflammatory and antioxidant effects of acetyl-L-carnitine on atherosclerotic rats. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e920250–e920251. [Google Scholar] [CrossRef]
- Tufekci, O.; Gunes, D.; Özoğul, C.; Kolatan, E.; Altun, Z.; Yılmaz, O.; Aktaş, S.; Erbayraktar, Z.; Kırkım, G.; Mutafoğlu, K. Evaluation of the effect of acetyl L-carnitine on experimental cisplatin nephrotoxicity. Chemotherapy 2009, 55, 451–459. [Google Scholar] [CrossRef]
- Traina, G. The neurobiology of acetyl-L-carnitine. Front. Biosci.-Landmark 2016, 21, 1314–1329. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.P.; Yang, X.Z.; Cao, G.P. Acetyl-l-carnitine prevents homocysteine-induced suppression of Nrf2/Keap1 mediated antioxidation in human lens epithelial cells. Mol. Med. Rep. 2015, 12, 1145–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepand, M.R.; Razavi-Azarkhiavi, K.; Omidi, A.; Zirak, M.R.; Sabzevari, S.; Kazemi, A.R.; Sabzevari, O. Effect of acetyl-l-carnitine on antioxidant status, lipid peroxidation, and oxidative damage of arsenic in rat. Biol. Trace Elem. Res. 2016, 171, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N. Delaying the mitochondrial decay of aging. Ann. N. Y. Acad. Sci. 2004, 1019, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Gargante, M.P.; Cristaldi, E.; Vacante, M.; Risino, C.; Cammalleri, L.; Pennisi, G.; Rampello, L. Acetyl-L-carnitine treatment in minimal hepatic encephalopathy. Dig. Dis. Sci. 2008, 53, 3018–3025. [Google Scholar] [CrossRef] [PubMed]
- Sima, A.A. Acetyl-L-Carnitine in Diabetic Polyneuropathy. CNS Drugs 2007, 21, 13–23. [Google Scholar] [CrossRef]
- Traina, G.; Bernardi, R.; Cataldo, E.; Macchi, M.; Durante, M.; Brunelli, M. In the rat brain acetyl-L-carnitine treatment modulates the expression of genes involved in neuronal ceroid lipofuscinosis. Mol. Neurobiol. 2008, 38, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Head, E.; Gharib, A.M.; Yuan, W.; Ingersoll, R.T.; Hagen, T.M.; Cotman, C.W.; Ames, B.N. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: Partial reversal by feeding acetyl-L-carnitine and/or R-α-lipoic acid. Proc. Natl. Acad. Sci. USA 2002, 99, 2356–2361. [Google Scholar] [CrossRef] [Green Version]
- Hagen, T.M.; Liu, J.; Lykkesfeldt, J.; Wehr, C.M.; Ingersoll, R.T.; Vinarsky, V.; Bartholomew, J.C.; Ames, B.N. Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc. Natl. Acad. Sci. USA 2002, 99, 1870–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elimrani, I.; Dionne, S.; Saragosti, D.; Qureshi, I.; Levy, E.; Delvin, E.; Seidman, E.G. Acetylcarnitine potentiates the anticarcinogenic effects of butyrate on SW480 colon cancer cells. Int. J. Oncol. 2015, 47, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Baci, D.; Bruno, A.; Bassani, B.; Tramacere, M.; Mortara, L.; Albini, A.; Noonan, D.M. Acetyl-l-carnitine is an anti-angiogenic agent targeting the VEGFR2 and CXCR4 pathways. Cancer Lett. 2018, 429, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Engle, D.B.; Belisle, J.A.; Gubbels, J.A.; Petrie, S.E.; Hutson, P.R.; Kushner, D.M.; Patankar, M.S. Effect of acetyl-l-carnitine on ovarian cancer cells’ proliferation, nerve growth factor receptor (Trk-A and p75) expression, and the cytotoxic potential of paclitaxel and carboplatin. Gynecol. Oncol. 2009, 112, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Gerharz, M.; Baranowsky, A.; Siebolts, U.; Eming, S.; Nischt, R.; Krieg, T.; Wickenhauser, C. Morphometric analysis of murine skin wound healing: Standardization of experimental procedures and impact of an advanced multitissue array technique. Wound Repair Regen. 2007, 15, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Sartorelli, A. Cancer chemotherapy. In Lange’s Basic and Clinical Pharmacology; McGraw Hill: New York, NY, USA, 2018; pp. 948–976. [Google Scholar]
- Carr, C.; Ng, J.; Wigmore, T. The side effects of chemotherapeutic agents. Curr. Anaesth. Crit. Care 2008, 19, 70–79. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, Y.-F. Natural compounds as anticancer agents: Experimental evidence. World J. Exp. Med. 2012, 2, 45. [Google Scholar] [CrossRef]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in cancer treatment: From preclinical studies to clinical practice. Front. Pharmacol. 2020, 10, 1614. [Google Scholar] [CrossRef] [Green Version]
- Momenzadeh, M.; Aria, A.; Ghadimi, K.; Moghaddas, A. Acetyl-L-carnitine for the prevention of taxane-induced neuropathy in patients with breast cancer: A systematic review and meta-analysis. Res. Pharm. Sci. 2023, 18, 112. [Google Scholar]
- Pillich, R.T.; Scarsella, G.; Risuleo, G. Reduction of apoptosis through the mitochondrial pathway by the administration of acetyl-L-carnitine to mouse fibroblasts in culture. Exp. Cell Res. 2005, 306, 1–8. [Google Scholar] [CrossRef]
- Bodaghi-Namileh, V.; Sepand, M.R.; Omidi, A.; Aghsami, M.; Seyednejad, S.A.; Kasirzadeh, S.; Sabzevari, O. Acetyl-l-carnitine attenuates arsenic-induced liver injury by abrogation of mitochondrial dysfunction, inflammation, and apoptosis in rats. Environ. Toxicol. Pharmacol. 2018, 58, 11–20. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, M.; Li, Q.; Zhao, H.; Wang, J.; Li, Y. Acetyl-l-carnitine inhibits TNF-α-induced insulin resistance via AMPK pathway in rat skeletal muscle cells. FEBS Lett. 2009, 583, 470–474. [Google Scholar] [CrossRef] [Green Version]
- Pisano, C.; Pratesi, G.; Laccabue, D.; Zunino, F.; Lo Giudice, P.; Bellucci, A.; Pacifici, L.; Camerini, B.; Vesci, L.; Castorina, M. Paclitaxel and Cisplatin-induced neurotoxicity: A protective role of acetyl-L-carnitine. Clin. Cancer Res. 2003, 9, 5756–5767. [Google Scholar] [PubMed]
- Pomponi, M.G.; Neri, G. Butyrate and acetyl-carnitine inhibit the cytogenetic expression of the fragile X in vitro. Am. J. Med. Genet. 1994, 51, 447–450. [Google Scholar] [CrossRef]
- Te Boekhorst, V.; Preziosi, L.; Friedl, P. Plasticity of cell migration in vivo and in silico. Annu. Rev. Cell Dev. Biol. 2016, 32, 491–526. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Nickel, A.; Daniel, H. Increased mitochondrial palmitoylcarnitine/carnitine countertransport by flavone causes oxidative stress and apoptosis in colon cancer cells. Cell. Mol. Life Sci. CMLS 2005, 62, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.; Nickel, A.; Daniel, H. Increased carnitine-dependent fatty acid uptake into mitochondria of human colon cancer cells induces apoptosis. J. Nutr. 2005, 135, 1510–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Zhang, Z.; Zhang, Y.; Pan, X.; Yu, L.; Liu, S. L-Carnitine ameliorates cancer cachexia in mice partly via the carnitine palmitoyltransferase-associated PPAR-γ signaling pathway. Oncol. Res. Treat. 2015, 38, 511–516. [Google Scholar] [CrossRef]
- Liu, S.; Wu, H.-J.; Zhang, Z.-Q.; Chen, Q.; Liu, B.; Wu, J.-P.; Zhu, L. L-carnitine ameliorates cancer cachexia in mice by regulating the expression and activity of carnitine palmityl transferase. Cancer Biol. Ther. 2011, 12, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roscilli, G.; Marra, E.; Mori, F.; Di Napoli, A.; Mancini, R.; Serlupi-Crescenzi, O.; Virmani, A.; Aurisicchio, L.; Ciliberto, G. Carnitines slow down tumor development of colon cancer in the DMH-chemical carcinogenesis mouse model. J. Cell. Biochem. 2013, 114, 1665–1673. [Google Scholar] [CrossRef]
- Montesano, A.; Senesi, P.; Vacante, F.; Mollica, G.; Benedini, S.; Mariotti, M.; Luzi, L.; Terruzzi, I. L-Carnitine counteracts in vitro fructose-induced hepatic steatosis through targeting oxidative stress markers. J. Endocrinol. Investig. 2020, 43, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.; McArthur, D.; Morris, A.; Williams, N. Cyclooxygenase-2 inhibition prevents migration of colorectal cancer cells to extracellular matrix by down-regulation of matrix metalloproteinase-2 expression. Dis. Colon Rectum 2008, 51, 342–347. [Google Scholar] [CrossRef]
- Lai, K.-C.; Huang, A.-C.; Hsu, S.-C.; Kuo, C.-L.; Yang, J.-S.; Wu, S.-H.; Chung, J.-G. Benzyl isothiocyanate (BITC) inhibits migration and invasion of human colon cancer HT29 cells by inhibiting matrix metalloproteinase-2/-9 and urokinase plasminogen (uPA) through PKC and MAPK signaling pathway. J. Agric. Food Chem. 2010, 58, 2935–2942. [Google Scholar] [CrossRef]
- Kurebayashi, J.; Otsuki, T.; Kunisue, H.; Mikami, Y.; Tanaka, K.; Yamamoto, S.; Sonoo, H. Expression of vascular endothelial growth factor (VEGF) family members in breast cancer. Jpn. J. Cancer Res. 1999, 90, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Arii, S.; Furutani, M.; Niwano, M.; Harada, T.; Mizumoto, M.; Mori, A.; Onodera, H.; Imamura, M. Predictive value of vascular endothelial growth factor (VEGF) in metastasis and prognosis of human colorectal cancer. Br. J. Cancer 1998, 78, 1379–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inai, T.; Mancuso, M.; Hashizume, H.; Baffert, F.; Haskell, A.; Baluk, P.; Hu-Lowe, D.D.; Shalinsky, D.R.; Thurston, G.; Yancopoulos, G.D. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am. J. Pathol. 2004, 165, 35–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albogami, S. The Potential Inhibitory Role of Acetyl-L-Carnitine on Proliferation, Migration, and Gene Expression in HepG2 and HT29 Human Adenocarcinoma Cell Lines. Curr. Issues Mol. Biol. 2023, 45, 2393-2408. https://doi.org/10.3390/cimb45030155
Albogami S. The Potential Inhibitory Role of Acetyl-L-Carnitine on Proliferation, Migration, and Gene Expression in HepG2 and HT29 Human Adenocarcinoma Cell Lines. Current Issues in Molecular Biology. 2023; 45(3):2393-2408. https://doi.org/10.3390/cimb45030155
Chicago/Turabian StyleAlbogami, Sarah. 2023. "The Potential Inhibitory Role of Acetyl-L-Carnitine on Proliferation, Migration, and Gene Expression in HepG2 and HT29 Human Adenocarcinoma Cell Lines" Current Issues in Molecular Biology 45, no. 3: 2393-2408. https://doi.org/10.3390/cimb45030155
APA StyleAlbogami, S. (2023). The Potential Inhibitory Role of Acetyl-L-Carnitine on Proliferation, Migration, and Gene Expression in HepG2 and HT29 Human Adenocarcinoma Cell Lines. Current Issues in Molecular Biology, 45(3), 2393-2408. https://doi.org/10.3390/cimb45030155