
A Novel Nonsense Pathogenic TTN Variant Identified in a Patient with Severe Dilated Cardiomyopathy

 ,
,  , , , , ,
, , , , , 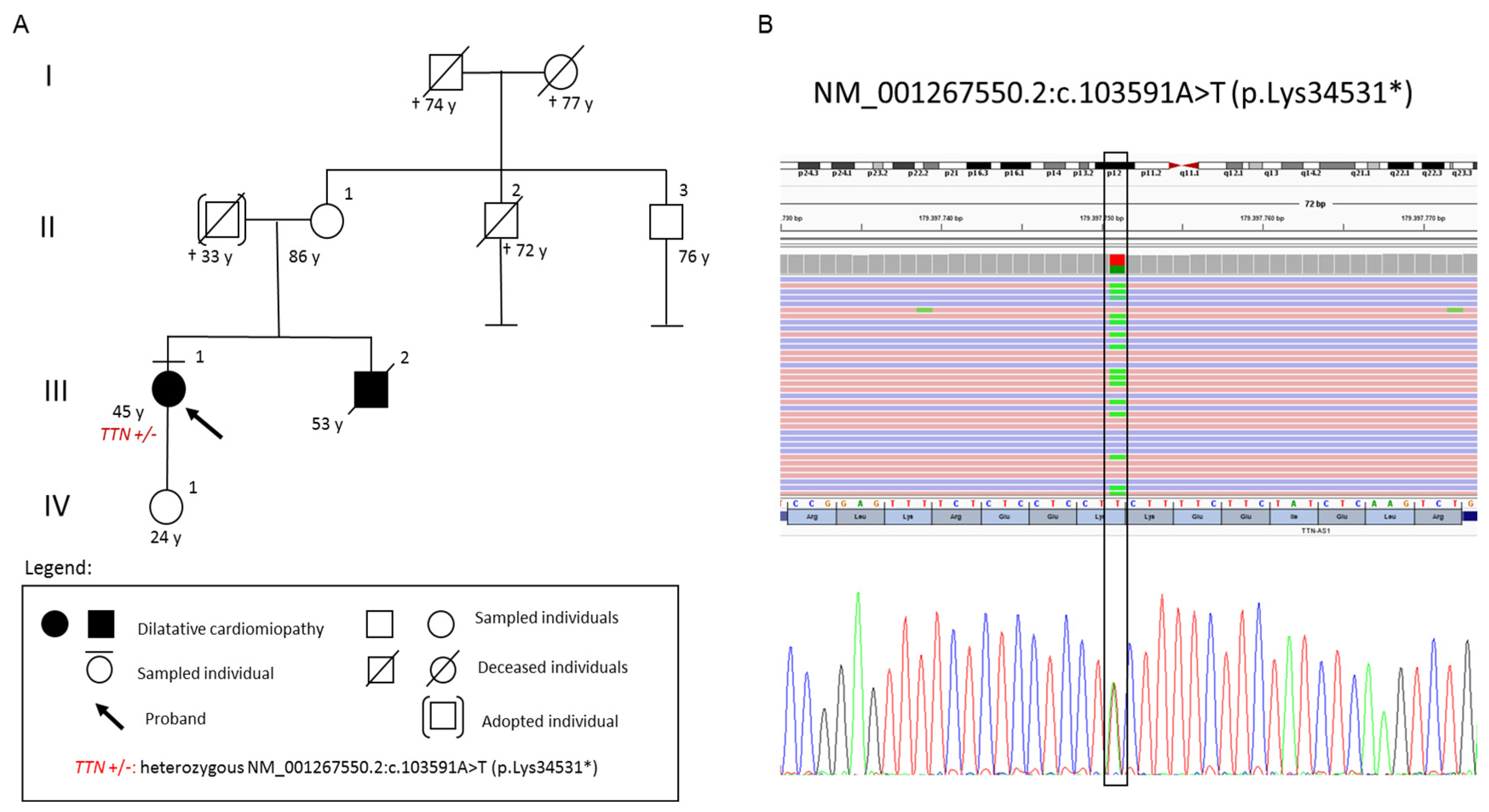{kind=link}
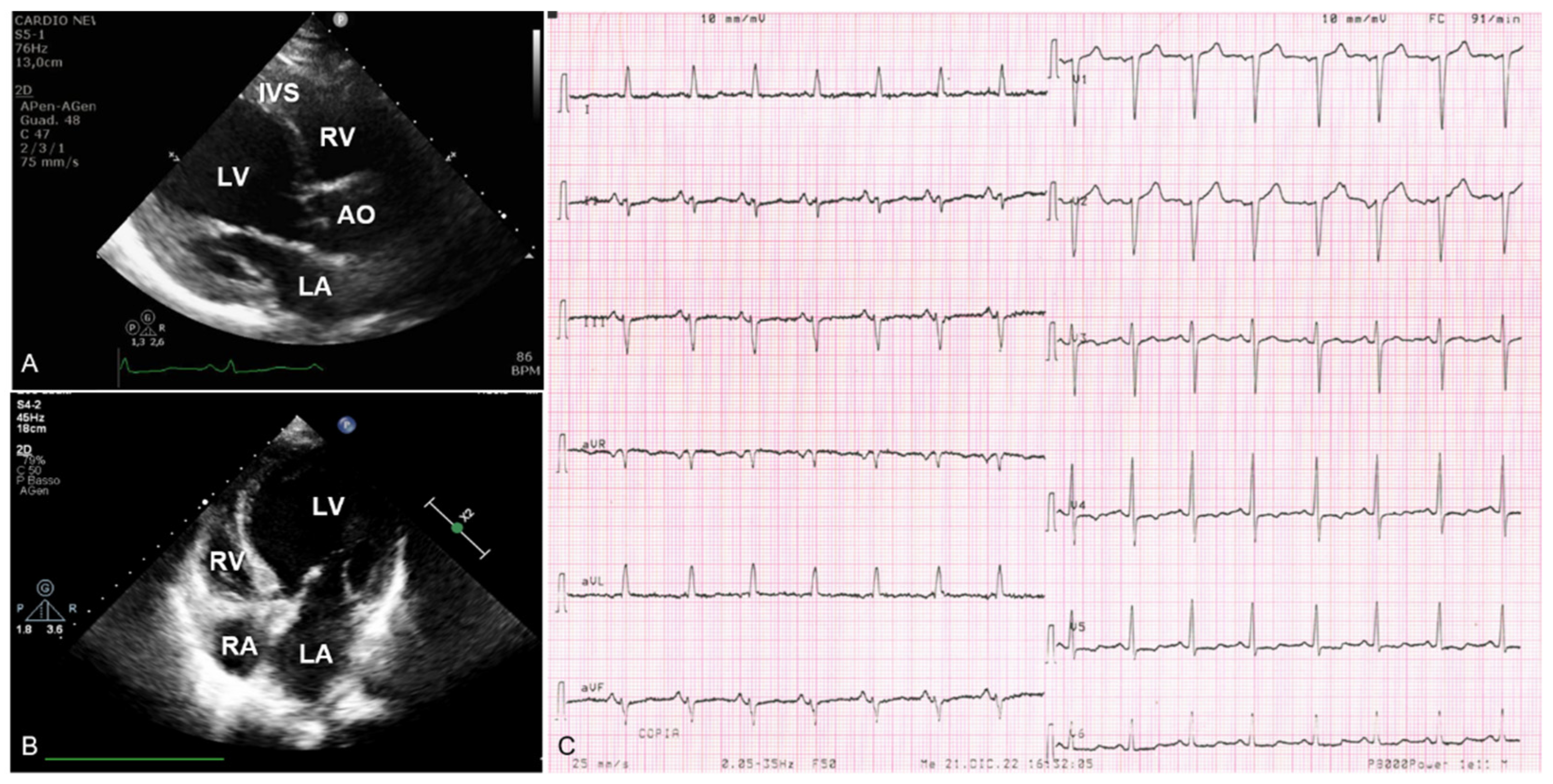{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Case Presentation
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated Cardiomyopathy: From Epidemiologic to Genetic Phenotypes: A Translational Review of Current Literature. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferies, J.L.; Towbin, J.A. Dilated Cardiomyopathy. Lancet 2010, 375, 752–762. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Jordan, E. Dilated Cardiomyopathy Overview. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Gaertner, A.; Bloebaum, J.; Brodehl, A.; Klauke, B.; Sielemann, K.; Kassner, A.; Fox, H.; Morshuis, M.; Tiesmeier, J.; Schulz, U.; et al. The Combined Human Genotype of Truncating and Mutations Is Associated with Severe and Early Onset of Dilated Cardiomyopathy. Genes 2021, 12, 883. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitás, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, Encoding the Giant Muscle Filament Titin, Cause Familial Dilated Cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef]
- Keller, H.; Finsterer, J.; Steger, C.; Wexberg, P.; Gatterer, E.; Khazen, C.; Stix, G.; Gerull, B.; Höftberger, R.; Weidinger, F. Novel c.367_369del LMNA Mutation Manifesting as Severe Arrhythmias, Dilated Cardiomyopathy, and Myopathy. Heart Lung 2012, 41, 382–386. [Google Scholar] [CrossRef] [PubMed]
- de Frutos, F.; Ochoa, J.P.; Navarro-Peñalver, M.; Baas, A.; Bjerre, J.V.; Zorio, E.; Méndez, I.; Lorca, R.; Verdonschot, J.A.J.; García-Granja, P.E.; et al. Natural History of MYH7-Related Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 80, 1447–1461. [Google Scholar] [CrossRef] [PubMed]
- Begay, R.L.; Tharp, C.A.; Martin, A.; Graw, S.L.; Sinagra, G.; Miani, D.; Sweet, M.E.; Slavov, D.B.; Stafford, N.; Zeller, M.J.; et al. Gene Splice Mutations Cause Dilated Cardiomyopathy. JACC Basic Transl. Sci. 2016, 1, 344–359. [Google Scholar] [CrossRef] [Green Version]
- Arimura, T.; Ishikawa, T.; Nunoda, S.; Kawai, S.; Kimura, A. Dilated Cardiomyopathy-Associated BAG3 Mutations Impair Z-Disc Assembly and Enhance Sensitivity to Apoptosis in Cardiomyocytes. Hum. Mutat. 2011, 32, 1481–1491. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Pinto, J.R.; Parks, S.B.; Kushner, J.D.; Li, D.; Ludwigsen, S.; Cowan, J.; Morales, A.; Parvatiyar, M.S.; Potter, J.D. Clinical and Functional Characterization of TNNT2 Mutations Identified in Patients with Dilated Cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Gaertner, A.; Klauke, B.; Felski, E.; Kassner, A.; Brodehl, A.; Gerdes, D.; Stanasiuk, C.; Ebbinghaus, H.; Schulz, U.; Dubowy, K.-O.; et al. Cardiomyopathy-Associated Mutations in the RS Domain Affect Nuclear Localization of RBM20. Hum. Mutat. 2020, 41, 1931–1943. [Google Scholar] [CrossRef]
- McNair, W.P.; Ku, L.; Taylor, M.R.G.; Fain, P.R.; Dao, D.; Wolfel, E.; Mestroni, L. Familial Cardiomyopathy Registry Research Group SCN5A Mutation Associated with Dilated Cardiomyopathy, Conduction Disorder, and Arrhythmia. Circulation 2004, 110, 2163–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Dieding, M.; Biere, N.; Unger, A.; Klauke, B.; Walhorn, V.; Gummert, J.; Schulz, U.; Linke, W.A.; Gerull, B.; et al. Functional Characterization of the Novel DES Mutation p.L136P Associated with Dilated Cardiomyopathy Reveals a Dominant Filament Assembly Defect. J. Mol. Cell. Cardiol. 2016, 91, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.P.; Kamisago, M.; Asahi, M.; Li, G.H.; Ahmad, F.; Mende, U.; Kranias, E.G.; MacLennan, D.H.; Seidman, J.G.; Seidman, C.E. Dilated Cardiomyopathy and Heart Failure Caused by a Mutation in Phospholamban. Science 2003, 299, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.R.; Siegfried, J.D.; Parvatiyar, M.S.; Li, D.; Norton, N.; Jones, M.A.; Liang, J.; Potter, J.D.; Hershberger, R.E. Functional Characterization of TNNC1 Rare Variants Identified in Dilated Cardiomyopathy. J. Biol. Chem. 2011, 286, 34404–34412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauveau, C.; Rowell, J.; Ferreiro, A. A Rising Titan: TTN Review and Mutation Update. Hum. Mutat. 2014, 35, 1046–1059. [Google Scholar] [CrossRef]
- Itoh-Satoh, M.; Hayashi, T.; Nishi, H.; Koga, Y.; Arimura, T.; Koyanagi, T.; Takahashi, M.; Hohda, S.; Ueda, K.; Nouchi, T.; et al. Titin Mutations as the Molecular Basis for Dilated Cardiomyopathy. Biochem. Biophys. Res. Commun. 2002, 291, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Bharmal, S.J.; Esbona, K.; Greaser, M.L. Titin Diversity—Alternative Splicing Gone Wild. J. Biomed. Biotechnol. 2010, 2010, 753675. [Google Scholar] [CrossRef] [Green Version]
- Eldemire, R.; Tharp, C.A.; Taylor, M.R.G.; Sbaizero, O.; Mestroni, L. The Sarcomeric Spring Protein Titin: Biophysical Properties, Molecular Mechanisms, and Genetic Mutations Associated with Heart Failure and Cardiomyopathy. Curr. Cardiol. Rep. 2021, 23, 121. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2018, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated Cardiomyopathy: The Complexity of a Diverse Genetic Architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Chmielewski, P.; Truszkowska, G.; Kowalik, I.; Rydzanicz, M.; Michalak, E.; Sobieszczańska-Małek, M.; Franaszczyk, M.; Stawiński, P.; Stępień-Wojno, M.; Oręziak, A.; et al. Titin-Related Dilated Cardiomyopathy: The Clinical Trajectory and the Role of Circulating Biomarkers in the Clinical Assessment. Diagnostics 2021, 12, 13. [Google Scholar] [CrossRef]
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.G.; Granzier, H.; Mestroni, L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated Allelic, Transcriptional, and Phenomic Dissection of the Cardiac Effects of Titin Truncations in Health and Disease. Sci. Transl. Med. 2015, 7, 270ra6. [Google Scholar] [CrossRef] [Green Version]
- Schafer, S.; de Marvao, A.; Adami, E.; Fiedler, L.R.; Ng, B.; Khin, E.; Rackham, O.J.L.; van Heesch, S.; Pua, C.J.; Kui, M.; et al. Titin-Truncating Variants Affect Heart Function in Disease Cohorts and the General Population. Nat. Genet. 2017, 49, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Akinrinade, O.; Koskenvuo, J.W.; Alastalo, T.-P. Prevalence of Titin Truncating Variants in General Population. PLoS ONE 2015, 10, e0145284. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, M.M.; Lorenzini, M.; Cicerchia, M.; Ochoa, J.P.; Hey, T.M.; Sabater Molina, M.; Restrepo-Cordoba, M.A.; Dal Ferro, M.; Stolfo, D.; Johnson, R.; et al. Clinical Phenotypes and Prognosis of Dilated Cardiomyopathy Caused by Truncating Variants in the TTN Gene. Circ. Heart Fail. 2020, 13, e006832. [Google Scholar] [CrossRef]
- Tayal, U.; Newsome, S.; Buchan, R.; Whiffin, N.; Halliday, B.; Lota, A.; Roberts, A.; Baksi, A.J.; Voges, I.; Midwinter, W.; et al. Phenotype and Clinical Outcomes of Titin Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 2264–2274. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Hazebroek, M.R.; Derks, K.W.J.; Barandiarán Aizpurua, A.; Merken, J.J.; Wang, P.; Bierau, J.; van den Wijngaard, A.; Schalla, S.M.; Abdul Hamid, M.A.; et al. Titin Cardiomyopathy Leads to Altered Mitochondrial Energetics, Increased Fibrosis and Long-Term Life-Threatening Arrhythmias. Eur. Heart J. 2018, 39, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Jansweijer, J.A.; Nieuwhof, K.; Russo, F.; Hoorntje, E.T.; Jongbloed, J.D.H.; Lekanne Deprez, R.H.; Postma, A.V.; Bronk, M.; van Rijsingen, I.A.W.; de Haij, S.; et al. Truncating Titin Mutations Are Associated with a Mild and Treatable Form of Dilated Cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Vissing, C.R.; Rasmussen, T.B.; Dybro, A.M.; Olesen, M.S.; Pedersen, L.N.; Jensen, M.; Bundgaard, H.; Christensen, A.H. Dilated Cardiomyopathy Caused by Truncating Titin Variants: Long-Term Outcomes, Arrhythmias, Response to Treatment and Sex Differences. J. Med. Genet. 2021, 58, 832–841. [Google Scholar] [CrossRef]
- Radke, M.H.; Polack, C.; Methawasin, M.; Fink, C.; Granzier, H.L.; Gotthardt, M. Deleting Full Length Titin Versus the Titin M-Band Region Leads to Differential Mechanosignaling and Cardiac Phenotypes. Circulation 2019, 139, 1813–1827. [Google Scholar] [CrossRef] [PubMed]
- Obermann, W.M.; Gautel, M.; Weber, K.; Fürst, D.O. Molecular Structure of the Sarcomeric M Band: Mapping of Titin and Myosin Binding Domains in Myomesin and the Identification of a Potential Regulatory Phosphorylation Site in Myomesin. EMBO J. 1997, 16, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchner, E.M.; Alexandrovich, A.; Kho, A.L.; Hensen, U.; Schäfer, L.V.; Brandmeier, B.; Gräter, F.; Grubmüller, H.; Gaub, H.E.; Gautel, M. Mechanoenzymatics of Titin Kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 13385–13390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeWinter, M.M.; Granzier, H.L. Titin Is a Major Human Disease Gene. Circulation 2013, 127, 938–944. [Google Scholar] [CrossRef] [Green Version]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The Landscape of Genetic Variation in Dilated Cardiomyopathy as Surveyed by Clinical DNA Sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Freiburg, A.; Gautel, M. A Molecular Map of the Interactions between Titin and Myosin-Binding Protein C. Implications for Sarcomeric Assembly in Familial Hypertrophic Cardiomyopathy. Eur. J. Biochem. 1996, 235, 317–323. [Google Scholar] [CrossRef]
- McAfee, Q.; Chen, C.Y.; Yang, Y.; Caporizzo, M.A.; Morley, M.; Babu, A.; Jeong, S.; Brandimarto, J.; Bedi, K.C., Jr.; Flam, E.; et al. Truncated Titin Proteins in Dilated Cardiomyopathy. Sci. Transl. Med. 2021, 13, eabd7287. [Google Scholar] [CrossRef] [PubMed]
- Fomin, A.; Gärtner, A.; Cyganek, L.; Tiburcy, M.; Tuleta, I.; Wellers, L.; Folsche, L.; Hobbach, A.J.; von Frieling-Salewsky, M.; Unger, A.; et al. Truncated Titin Proteins and Titin Haploinsufficiency Are Targets for Functional Recovery in Human Cardiomyopathy due to TTN Mutations. Sci. Transl. Med. 2021, 13, eabd3079. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Amor-Salamanca, A.; Tayal, U.; Govind, R.; Serrano, I.; Salazar-Mendiguchía, J.; García-Pinilla, J.M.; Pascual-Figal, D.A.; Nuñez, J.; Guzzo-Merello, G.; et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J. Am. Coll. Cardiol. 2018, 71, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Lota, A.S.; Hazebroek, M.R.; Theotokis, P.; Wassall, R.; Salmi, S.; Halliday, B.P.; Tayal, U.; Verdonschot, J.; Meena, D.; Owen, R.; et al. Genetic Architecture of Acute Myocarditis and the Overlap With Inherited Cardiomyopathy. Circulation 2022, 146, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Kim, Y.; Restrepo-Cordoba, M.A.; Lunde, I.G.; Wakimoto, H.; Smith, A.M.; Toepfer, C.N.; Getz, K.; Gorham, J.; Patel, P.; et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation 2019, 140, 31–41. [Google Scholar] [CrossRef]
- Ware, J.S.; Seidman, J.G.; Arany, Z. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N. Engl. J. Med. 2016, 374, 2601–2602. [Google Scholar] [CrossRef] [PubMed]
- Piriou, N.; Marteau, L.; Kyndt, F.; Serfaty, J.M.; Toquet, C.; Le Gloan, L.; Warin-Fresse, K.; Guijarro, D.; Le Tourneau, T.; Conan, E.; et al. Familial Screening in Case of Acute Myocarditis Reveals Inherited Arrhythmogenic Left Ventricular Cardiomyopathies. ESC Heart Fail. 2020, 7, 1520–1533. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micolonghi, C.; Fabiani, M.; Pagannone, E.; Savio, C.; Ricci, M.; Caroselli, S.; Gambioli, V.; Musumeci, B.; Germani, A.; Tini, G.; et al. A Novel Nonsense Pathogenic TTN Variant Identified in a Patient with Severe Dilated Cardiomyopathy. Curr. Issues Mol. Biol. 2023, 45, 2422-2430. https://doi.org/10.3390/cimb45030157
Micolonghi C, Fabiani M, Pagannone E, Savio C, Ricci M, Caroselli S, Gambioli V, Musumeci B, Germani A, Tini G, et al. A Novel Nonsense Pathogenic TTN Variant Identified in a Patient with Severe Dilated Cardiomyopathy. Current Issues in Molecular Biology. 2023; 45(3):2422-2430. https://doi.org/10.3390/cimb45030157
Chicago/Turabian StyleMicolonghi, Caterina, Marco Fabiani, Erika Pagannone, Camilla Savio, Marta Ricci, Silvia Caroselli, Vittoria Gambioli, Beatrice Musumeci, Aldo Germani, Giacomo Tini, and et al. 2023. "A Novel Nonsense Pathogenic TTN Variant Identified in a Patient with Severe Dilated Cardiomyopathy" Current Issues in Molecular Biology 45, no. 3: 2422-2430. https://doi.org/10.3390/cimb45030157
APA StyleMicolonghi, C., Fabiani, M., Pagannone, E., Savio, C., Ricci, M., Caroselli, S., Gambioli, V., Musumeci, B., Germani, A., Tini, G., Autore, C., Pizzuti, A., Visco, V., Rubattu, S., Petrucci, S., & Piane, M. (2023). A Novel Nonsense Pathogenic TTN Variant Identified in a Patient with Severe Dilated Cardiomyopathy. Current Issues in Molecular Biology, 45(3), 2422-2430. https://doi.org/10.3390/cimb45030157