
Study on DNA Storage Encoding Based IAOA under Innovation Constraints



Abstract
:1. Introduction
2. Algorithm Description
2.1. Arithmetic Optimization Algorithm
2.2. IAOA
2.2.1. Elementary Function Perturbation
2.2.2. Double Adaptive Weighting Strategy
| Algorithm 1. Pseudo-code of the IAOA. |
| 1: Initialization parameters and population location i (i = 1,2...N) 2: While(t < T) 3: 4: for i = 1 : N 5: for j = 1 : N 6: if r1 > MOA 7: if r2 > 0.5 (exploration phase) 8: 9: else 10: 11: end if 12: if r3 > 0.5 (development phase) 13: 14: else 15: 16: end if 17: end if 18: end for 19: end for 20: end while 21: Return to the optimal solution |
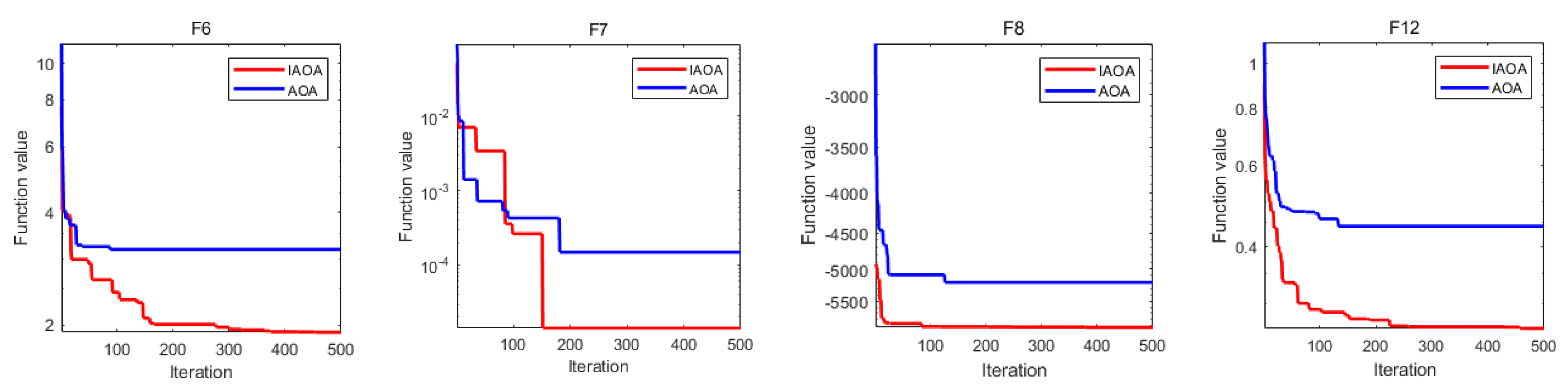
2.3. Benchmark Function Comparison
2.4. Wilcoxon Rank Sum Test
3. Construct DNA Storage Sets
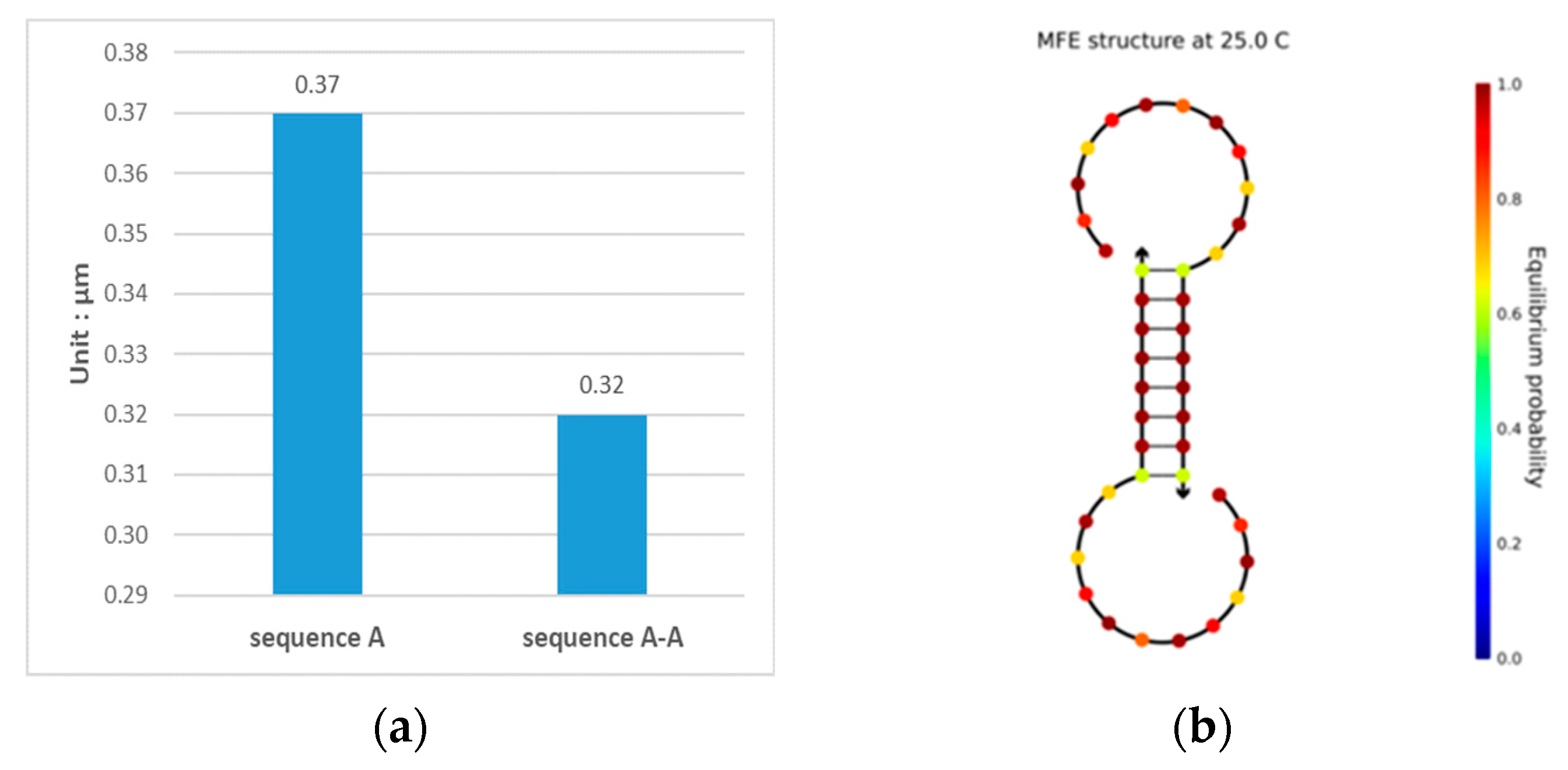
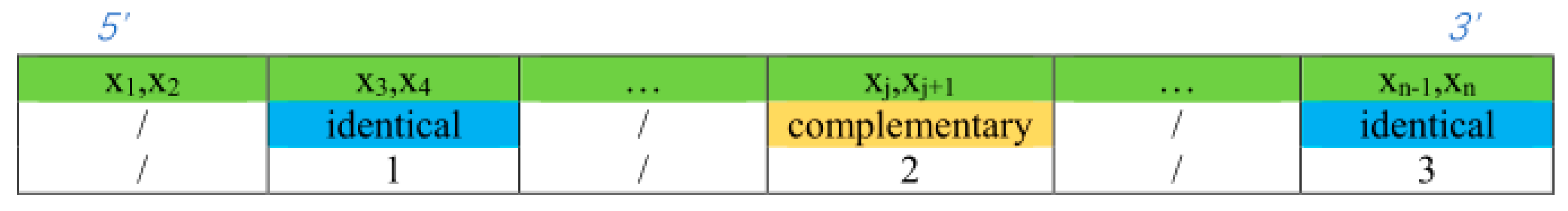
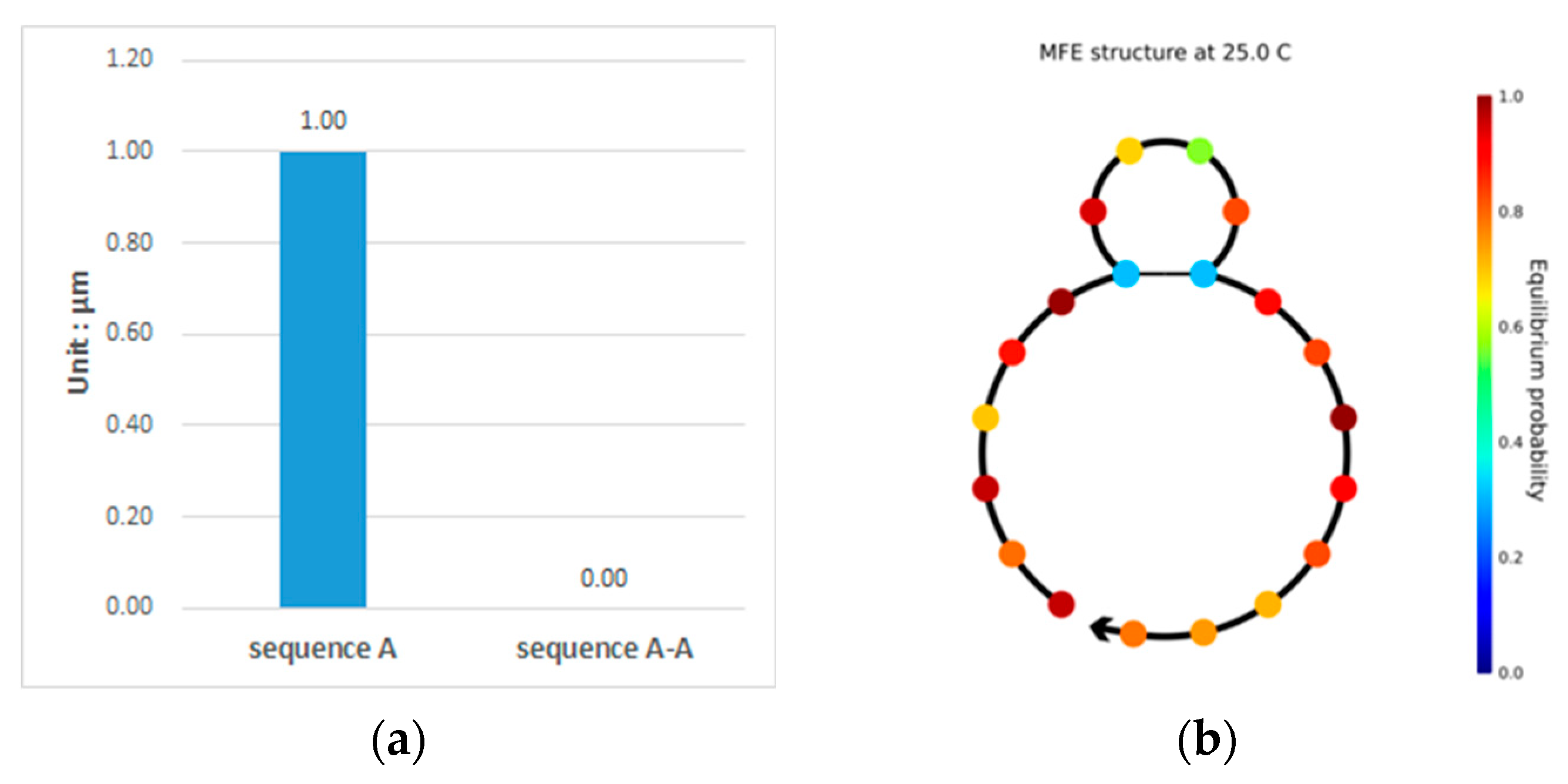
3.1. Double-Matching Constraint
3.2. Error-Pairing Constraint
3.3. Hamming Distance Constraint
3.4. GC Content Constraint
3.5. No-Runlength Constraint
3.6. DNA Storage Sets Constructed Using Traditional Combinatorial Constraints
3.7. Encoding Sets Constructed Using Double-Matching and Error-Pairing Constraints
3.8. Storage Set Quality Comparison
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dong, Y.; Sun, F.; Ping, Z.; Ouyang, Q.; Qian, L. DNA storage: Research landscape and future prospects. Natl. Sci. Rev. 2020, 7, 1092–1107. [Google Scholar] [CrossRef] [PubMed]
- Ceze, L.; Nivala, J.; Strauss, K. Molecular digital data storage using DNA. Nat. Rev. Genet. 2019, 20, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.H.F.; STOKES, A.R.; Wilson, H.R. Molecular structure of deoxypentose nucleic acids. Nature 1953, 171, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Ping, Z.; Ma, D.; Huang, X.; Chen, S.; Liu, L.; Guo, F.; Zhu, S.J.; Shen, Y. Carbon-based archiving: Current progress and future prospects of DNA-based data storage. GigaScience 2019, 8, giz075. [Google Scholar] [CrossRef]
- Erlich, Y.; Zielinski, D. DNA Fountain enables a robust and efficient storage architecture. Science 2017, 355, 950–954. [Google Scholar] [CrossRef]
- Church, G.M.; Gao, Y.; Kosuri, S. Next-generation digital information storage in DNA. Science 2012, 337, 1628. [Google Scholar] [CrossRef]
- Goldman, N.; Bertone, P.; Chen, S.; Dessimoz, C.; LeProust, E.M.; Sipos, B.; Birney, E. Towards practical, high-capacity, low-maintenance information storage in synthesized DNA. Nature 2013, 494, 77–80. [Google Scholar] [CrossRef]
- Tabatabaei Yazdi, S.; Yuan, Y.; Ma, J.; Zhao, H.; Milenkovic, O. A rewritable, random-access DNA-based storage system. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Hoshika, S.; Leal, N.A.; Kim, M.-J.; Kim, M.-S.; Karalkar, N.B.; Kim, H.-J.; Bates, A.M.; Watkins, N.E., Jr.; SantaLucia, H.A.; Meyer, A.J. Hachimoji DNA and RNA: A genetic system with eight building blocks. Science 2019, 363, 884–887. [Google Scholar] [CrossRef]
- Ping, Z.; Chen, S.; Zhou, G.; Huang, X.; Zhu, S.J.; Zhang, H.; Lee, H.H.; Lan, Z.; Cui, J.; Chen, T. Towards practical and robust DNA-based data archiving using the yin–yang codec system. Nat. Comput. Sci. 2022, 2, 234–242. [Google Scholar] [CrossRef]
- Choi, Y.; Ryu, T.; Lee, A.C.; Choi, H.; Lee, H.; Park, J.; Song, S.-H.; Kim, S.; Kim, H.; Park, W. High information capacity DNA-based data storage with augmented encoding characters using degenerate bases. Sci. Rep. 2019, 9, 6582. [Google Scholar] [CrossRef] [PubMed]
- Anavy, L.; Vaknin, I.; Atar, O.; Amit, R.; Yakhini, Z. Data storage in DNA with fewer synthesis cycles using composite DNA letters. Nat. Biotechnol. 2019, 37, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kong, L.; Wang, F.; Li, B.; Ma, C.; Chen, D.; Liu, K.; Fan, C.; Zhang, H. Information stored in nanoscale: Encoding data in a single DNA strand with Base64. Nano Today 2020, 33, 100871. [Google Scholar] [CrossRef]
- Ping, Z.; Zhang, H.; Chen, S.; Zhuang, Q.; Zhu, S.J.; Shen, Y. Chamaeleo: A robust library for DNA storage coding schemes. bioRxiv 2020, 2020, 892588. [Google Scholar]
- Cao, B.; Li, X.; Zhang, X.; Wang, B.; Zhang, Q.; Wei, X. Designing uncorrelated address constrain for DNA storage by DMVO algorithm. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, B.; Lv, H.; Yin, Q.; Zhang, Q.; Wei, X. Constraining DNA Sequences with a Triplet-Bases Unpaired. IEEE Trans. Nanobiosci. 2020, 19, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Shomorony, I.; Heckel, R. DNA-Based Storage: Models and Fundamental Limits. IEEE Trans. Inf. Theory 2021, 67, 3675–3689. [Google Scholar] [CrossRef]
- Li, M.; Wu, J.; Dai, J.; Jiang, Q.; Qu, Q.; Huang, X.; Wang, Y. A self-contained and self-explanatory DNA storage system. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Schwarz, P.M.; Freisleben, B. NOREC4DNA: Using near-optimal rateless erasure codes for DNA storage. BMC Bioinform. 2021, 22, 406. [Google Scholar] [CrossRef]
- Park, S.-J.; Lee, Y.; No, J.-S. Iterative DNA Coding Scheme with GC Balance and Run-Length Constraints Using a Greedy Algorithm. arXiv 2021, preprint. arXiv:2103.03540. [Google Scholar]
- Zheng, Y.; Wu, J.; Wang, B. CLGBO: An algorithm for constructing highly robust coding sets for DNA storage. Front. Genet. 2021, 12, 644945. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zheng, Y.; Wang, B.; Zhang, Q. Enhancing Physical and Thermodynamic Properties of DNA Storage Sets with End-Constraint. IEEE Trans. NanoBiosci. 2021, 21, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, S.; Zou, L. Design of DNA Storage Coding with Enhanced Constraints. Entropy 2022, 24, 1151. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, Y.; Liu, Y.; Wu, Q.; Su, J.; Wang, F.; Chen, D.; Fan, C.; Liu, K.; Zhang, H. DNA-Based Concatenated Encoding System for High-Reliability and High-Density Data Storage. Small Methods 2022, 6, 2101335. [Google Scholar] [CrossRef] [PubMed]
- Abualigah, L.; Diabat, A.; Mirjalili, S.; Abd Elaziz, M.; Gandomi, A.H. The arithmetic optimization algorithm. Comput. Methods Appl. Mech. Eng. 2021, 376, 113609. [Google Scholar] [CrossRef]
- Wang, R.-B.; Wang, W.-F.; Xu, L.; Pan, J.-S.; Chu, S.-C. An adaptive parallel arithmetic optimization algorithm for robot path planning. J. Adv. Transp. 2021, 2021, 3606895. [Google Scholar] [CrossRef]
- Agushaka, J.O.; Ezugwu, A.E. Advanced arithmetic optimization algorithm for solving mechanical engineering design problems. PLoS ONE 2021, 16, e0255703. [Google Scholar] [CrossRef]
- Hao, W.-K.; Wang, J.-S.; Li, X.-D.; Wang, M.; Zhang, M. Arithmetic optimization algorithm based on elementary function disturbance for solving economic load dispatch problem in power system. Appl. Intell. 2022, 52, 11846–11872. [Google Scholar] [CrossRef]
- Abualigah, L.; Diabat, A.; Sumari, P.; Gandomi, A.H. A novel evolutionary arithmetic optimization algorithm for multilevel thresholding segmentation of COVID-19 ct images. Processes 2021, 9, 1155. [Google Scholar] [CrossRef]
- Shan, W.; Qiao, Z.; Heidari, A.A.; Chen, H.; Turabieh, H.; Teng, Y. Double adaptive weights for stabilization of moth flame optimizer: Balance analysis, engineering cases, and medical diagnosis. Knowl. Based Syst. 2021, 214, 106728. [Google Scholar] [CrossRef]
- Molga, M.; Smutnicki, C. Test functions for optimization needs. Test Funct. Optim. Needs 2005, 101, 48. [Google Scholar]
- Mirjalili, S.; Mirjalili, S.M.; Hatamlou, A. Multi-verse optimizer: A nature-inspired algorithm for global optimization. Neural Comput. Appl. 2016, 27, 495–513. [Google Scholar] [CrossRef]
- Rashedi, E.; Nezamabadi-Pour, H.; Saryazdi, S. GSA: A gravitational search algorithm. Inf. Sci. 2009, 179, 2232–2248. [Google Scholar] [CrossRef]
- Mirjalili, S.; Lewis, A. The Whale Optimization Algorithm. Adv. Eng. Softw. 2016, 95, 51–67. [Google Scholar] [CrossRef]
- Mirjalili, S.; Gandomi, A.H.; Mirjalili, S.Z.; Saremi, S.; Faris, H.; Mirjalili, S.M. Salp Swarm Algorithm: A bio-inspired optimizer for engineering design problems. Adv. Eng. Softw. 2017, 114, 163–191. [Google Scholar] [CrossRef]
- Mirjalili, S. SCA: A sine cosine algorithm for solving optimization problems. Knowl. Based Syst. 2016, 96, 120–133. [Google Scholar] [CrossRef]
- Zheng, R.; Jia, H.; Abualigah, L.; Liu, Q.; Wang, S. Deep ensemble of slime mold algorithm and arithmetic optimization algorithm for global optimization. Processes 2021, 9, 1774. [Google Scholar] [CrossRef]
- Derrac, J.; García, S.; Molina, D.; Herrera, F. A practical tutorial on the use of nonparametric statistical tests as a methodology for comparing evolutionary and swarm intelligence algorithms. Swarm Evol. Comput. 2011, 1, 3–18. [Google Scholar] [CrossRef]
- Chaves-González, J.M.; Martínez-Gil, J. An efficient design for a multi-objective evolutionary algorithm to generate DNA libraries suitable for computation. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 542–558. [Google Scholar] [CrossRef]
- Kwok, S.; Kellogg, D.; McKinney, N.; Spasic, D.; Goda, L.; Levenson, C.; Sninsky, J. Effects of primer-template mismatches on the polymerase chain reaction: Human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 1990, 18, 999–1005. [Google Scholar] [CrossRef]
- Aboluion, N.; Smith, D.H.; Perkins, S. Linear and nonlinear constructions of DNA codes with Hamming distance d, constant GC-content and a reverse-complement constraint. Discret. Math. 2012, 312, 1062–1075. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Q.; Wei, X. Tabu variable neighborhood search for designing DNA barcodes. IEEE Trans. NanoBiosci. 2019, 19, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Limbachiya, D.; Gupta, M.K.; Aggarwal, V. Family of constrained codes for archival DNA data storage. IEEE Commun. Lett. 2018, 22, 1972–1975. [Google Scholar] [CrossRef]
- Yin, Q.; Cao, B.; Li, X.; Wang, B.; Zhang, Q.; Wei, X. An intelligent optimization algorithm for constructing a DNA storage code: NOL-HHO. Int. J. Mol. Sci. 2020, 21, 2191. [Google Scholar] [CrossRef] [PubMed]
- Xiaoru, L.; Ling, G. Combinatorial constraint coding based on the EORS algorithm in DNA storage. PLoS ONE 2021, 16, e0255376. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.; Stefanovic, D. Designing nucleotide sequences for computation: A survey of constraints. Lect. Notes Comput. Sci. 2006, 3892, 275–289. [Google Scholar]
- Yang, G.; Wang, B.; Zheng, X.; Zhou, C.; Zhang, Q. IWO algorithm based on niche crowding for DNA sequence design. Interdiscip. Sci. Comput. Life Sci. 2017, 9, 341–349. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Metric | IAOA | EAOA | DAOA | pAOA | AOA | SCA | SSA | WOA | GSA | MVO |
|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | AVG | 0 | 8.06 × 10−6 | 0 | 7.17 × 10−7 | 8.65 × 10−26 | 7.6776 | 1.58 × 10−7 | 1.41 × 10−30 | 2.53 × 10−16 | 1.34 × 100 |
| STD | 0 | 2.88 × 10−5 | 0 | 1.87 × 10−6 | 4.74 × 10−25 | 12.3019 | 1.71 × 10−7 | 4.91 × 10−30 | 9.67 × 10−17 | 5.38 × 10−1 | |
| F2 | AVG | 0 | 9.93 × 10−98 | 0 | 7.74 × 10−70 | 0 | 0.01806 | 2.66293 | 1.06 × 10−21 | 0.055655 | 2.20 × 100 |
| STD | 0 | 5.44 × 10−97 | 0 | 4.24 × 10−69 | 0 | 0.02457 | 1.66802 | 2.39 × 10−21 | 0.194074 | 7.31 × 100 | |
| F3 | AVG | 0 | 0.002257 | 0 | 0.002124 | 0.008014 | 9961.453 | 1709.94 | 5.39 × 10−7 | 896.5347 | 2.04 × 102 |
| STD | 0 | 0.002291 | 0 | 0.00177 | 0.01192 | 6699.979 | 11242.3 | 2.93 × 10−6 | 318.9559 | 6.63 × 101 | |
| F4 | AVG | 0 | 0.012309 | 0 | 0.008175 | 0.02667 | 36.7941 | 11.6741 | 0.072581 | 7.35487 | 2.16 × 100 |
| STD | 0 | 0.004386 | 0 | 0.003422 | 0.02021 | 13.1414 | 4.1792 | 0.39747 | 1.741452 | 8.66 × 10−1 | |
| F5 | AVG | 27.7041 | 28.5909 | 28.0863 | 28.5339 | 28.3946 | 27188.68 | 296.125 | 27.86558 | 2.84 × 101 | 7.89 × 102 |
| STD | 0.27902 | 0.29615 | 0.34232 | 0.13585 | 0.3301 | 72171.04 | 508.863 | 0.763626 | 2.00 × 10−1 | 8.74 × 102 | |
| F6 | AVG | 1.937 | 2.4126 | 4.8677 | 1.5769 | 3.2316 | 21.998 | 1.80 × 10−7 | 3.116266 | 2.50 × 10−16 | 1.34 × 100 |
| STD | 0.38153 | 0.44494 | 0.50584 | 0.2591 | 0.2455 | 27.8352 | 3.00 × 10−7 | 0.532429 | 1.74 × 10−16 | 3.43 × 10−1 | |
| F7 | AVG | 5.42 × 10−5 | 6.25 × 10−5 | 7.40 × 10−5 | 4.51 × 10−5 | 5.45 × 10−5 | 0.08458 | 0.1757 | 0.001425 | 0.089441 | 3.21 × 10−2 |
| STD | 7.85 × 10−5 | 8.45 × 10−5 | 8.20 × 10−5 | 3.70 × 10−5 | 5.15 × 10−5 | 0.09798 | 0.0629 | 0.001149 | 0.04339 | 1.32 × 10−2 |
| ID | Metric | IAOA | EAOA | DAOA | pAOA | AOA | SCA | SSA | WOA | GSA | MVO |
|---|---|---|---|---|---|---|---|---|---|---|---|
| F8 | AVG | −6717.1783 | −6021.7325 | −4156.4559 | −6953.88 | −5395.427 | −3771.665 | −7455.8 | −5080.76 | −2821.07 | −7550 |
| STD | 580.0765 | 698.8845 | 599.9788 | 424.3747 | 436.8011 | 293.4553 | 772.811 | 695.7968 | 493.0375 | 6.27 × 102 | |
| F9 | AVG | 0 | 0 | 0 | 0 | 0 | 41.6519 | 58.3708 | 0 | 25.96841 | 1.20 × 102 |
| STD | 0 | 0 | 0 | 0 | 0 | 44.3312 | 20.016 | 0 | 7.470068 | 3.29 × 101 | |
| F10 | AVG | 8.88 × 10−16 | 8.88 × 10−16 | 8.88 × 10−16 | 8.88 × 10−16 | 8.88 × 10−16 | 14.2857 | 2.6796 | 7.4043 | 0.062087 | 2.03 × 100 |
| STD | 0 | 0 | 0 | 0 | 0 | 8.5929 | 0.8275 | 9.897572 | 0.23628 | 5.47 × 10−1 | |
| F11 | AVG | 0 | 0.004063 | 0 | 1.31 × 10−5 | 0.1689 | 0.8665 | 0.016 | 0.000289 | 27.70154 | 8.60 × 10−1 |
| STD | 0 | 0.009628 | 0 | 8.60 × 10−6 | 0.1347 | 0.3957 | 0.0112 | 0.001586 | 5.040343 | 8.21 × 10−2 | |
| F12 | AVG | 0.20433 | 0.5004 | 0.70028 | 0.2241 | 0.5195 | 183961.2 | 6.9915 | 0.339676 | 1.799617 | 2.43 × 100 |
| STD | 0.034199 | 0.043832 | 0.066711 | 0.0416 | 0.04741 | 841708.7 | 4.4175 | 0.214864 | 0.95114 | 1.39 × 100 | |
| F13 | AVG | 2.2663 | 2.7269 | 2.7643 | 2.7669 | 2.8475 | 109173.3 | 15.8757 | 1.889015 | 8.899084 | 1.96 × 10−1 |
| STD | 0.24611 | 0.30181 | 0.298724 | 0.1274 | 0.07852 | 266184.2 | 16.1462 | 0.266088 | 7.126241 | 1.26 × 10−1 |
| Comparison | p-Value |
|---|---|
| IAOA-AOA | 0.005847 |
| IAOA-pAOA | 0.028056 |
| IAOA-SCA | 0.001474 |
| IAOA-SSA | 0.039243 |
| IAOA-WOA | 0.026231 |
| IAOA-GSA | 0.009633 |
| IAOA-MVO | 0.008775 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|
| Altruistic | 11 | |||||||
| 4 | NOL-HHO | 12 | ||||||
| IAOA | 12 | |||||||
| Altruistic | 17 | 7 | ||||||
| 5 | NOL-HHO | 20 | 8 | |||||
| IAOA | 20 | 8 | ||||||
| Altruistic | 44 | 16 | 6 | |||||
| 6 | NOL-HHO | 55 | 23 | 8 | ||||
| IAOA | 61 | 24 | 8 | |||||
| Altruistic | 110 | 36 | 11 | 4 | ||||
| 7 | NOL-HHO | 121 | 42 | 14 | 7 | |||
| IAOA | 136 | 46 | 16 | 7 | ||||
| Altruistic | 289 | 86 | 29 | 9 | 4 | |||
| 8 | NOL-HHO | 339 | 108 | 35 | 13 | 5 | ||
| IAOA | 373 | 114 | 39 | 16 | 5 | |||
| Altruistic | 662 | 199 | 59 | 15 | 8 | 4 | ||
| 9 | NOL-HHO | 705 | 216 | 69 | 22 | 11 | 4 | |
| IAOA | 789 | 231 | 71 | 27 | 11 | 5 | ||
| Altruistic | 1810 | 525 | 141 | 43 | 7 | 5 | 4 | |
| 10 | NOL-HHO | 1796 | 546 | 148 | 51 | 20 | 9 | 4 |
| IAOA | 1945 | 549 | 156 | 56 | 22 | 10 | 5 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|
| 4 | 12 | ||||||
| 5 | 20 | 8 | |||||
| 6 | 58 | 22 | 8 | ||||
| 7 | 125 | 42 | 17 | 6 | |||
| 8 | 322 | 96 | 29 | 13 | 5 | ||
| 9 | 587 | 194 | 50 | 18 | 9 | 6 | |
| 10 | 1206 | 398 | 117 | 46 | 16 | 8 | 4 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|
| 4 | 12 | ||||||
| 5 | 20 | 8 | |||||
| 6 | 60 | 23 | 8 | ||||
| 7 | 126 | 43 | 18 | 6 | |||
| 8 | 338 | 119 | 33 | 14 | 5 | ||
| 9 | 598 | 201 | 58 | 21 | 11 | 7 | |
| 10 | 1391 | 408 | 126 | 49 | 18 | 8 | 4 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|
| 4 | 12 | ||||||
| 5 | 20 | 8 | |||||
| 6 | 45 | 17 | 7 | ||||
| 7 | 124 | 42 | 15 | 6 | |||
| 8 | 245 | 79 | 28 | 11 | 5 | ||
| 9 | 577 | 178 | 54 | 19 | 9 | 4 | |
| 10 | 1073 | 374 | 110 | 39 | 15 | 8 | 4 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|
| 8 | 3.5538 | 4.0477 | 6.0929 | 5.4760 | 4.6825 | |||
| 3.0968 | 3.3164 | 5.3873 | 5.1429 | 4.3069 | ||||
| 9 | 3.3585 | 2.6020 | 5.6688 | 4.6172 | 4.5196 | 4.8333 | ||
| 2.7969 | 2.5061 | 4.3512 | 4.1070 | 4.2980 | 4.7471 | |||
| 10 | 6.8984 | 2.9266 | 3.3843 | 3.2426 | 4.3286 | 2.6257 | 3.0167 | |
| 5.9291 | 2.7699 | 2.7484 | 2.8492 | 3.2602 | 2.5596 | 2.8941 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|
| 8 | 3.5538 | 4.0477 | 6.0929 | 5.4760 | 4.6825 | |||
| 3.0775 | 3.2602 | 5.1959 | 4.7633 | 3.9234 | ||||
| 9 | 3.3585 | 2.6020 | 5.6688 | 4.6172 | 4.5196 | 4.8333 | ||
| 2.7502 | 1.9650 | 4.0618 | 3.6314 | 3.9391 | 4.0712 | |||
| 10 | 6.8984 | 2.9266 | 3.3843 | 3.2426 | 4.3286 | 2.6257 | 3.0167 | |
| 5.1916 | 2.7484 | 2.4791 | 2.7502 | 3.3756 | 2.3892 | 2.8166 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|
| 8 | 3.5538 | 4.0477 | 6.0929 | 5.4760 | 4.6825 | |||
| 2.6499 | 3.4963 | 4.7498 | 4.4845 | 3.2870 | ||||
| 9 | 3.3585 | 2.6020 | 5.6688 | 4.6172 | 4.5196 | 4.8333 | ||
| 2.5067 | 1.4909 | 3.9142 | 3.1942 | 3.5079 | 3.8270 | |||
| 10 | 6.8984 | 2.9266 | 3.3843 | 3.2426 | 4.3286 | 2.6257 | 3.0167 | |
| 4.0036 | 2.2510 | 1.8384 | 2.5098 | 3.0063 | 2.2795 | 2.7492 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|
| 8 | 134 | 38 | 14 | 5 | 2 | |||
| 141 | 48 | 16 | 6 | 2 | ||||
| 9 | 751 | 251 | 68 | 25 | 13 | 7 | ||
| 776 | 258 | 81 | 29 | 16 | 8 | |||
| 10 | 3352 | 1064 | 335 | 128 | 46 | 19 | 18 | |
| 3832 | 1089 | 367 | 137 | 57 | 20 | 8 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|
| 8 | 0.4162 | 0.3958 | 0.4828 | 0.3846 | 0.4 | |||
| 0.4172 | 0.4034 | 0.4848 | 0.4286 | 0.4 | ||||
| 9 | 1.2794 | 1.2938 | 1.36 | 1.3889 | 1.4444 | 1.1667 | ||
| 1.2977 | 1.2835 | 1.3966 | 1.3810 | 1.4545 | 1.1429 | |||
| 10 | 2.7794 | 2.6734 | 2.8632 | 2.7826 | 2.875 | 2.375 | 2.25 | |
| 2.7549 | 2.6691 | 2.9127 | 2.7959 | 3.1667 | 2.5 | 2.75 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|
| 8 | 156 | 47 | 19 | 8 | 3 | |||
| 127 | 37 | 15 | 4 | 2 | ||||
| 9 | 1024 | 308 | 100 | 38 | 17 | 6 | ||
| 683 | 222 | 73 | 24 | 12 | 4 | |||
| 10 | 5412 | 1482 | 460 | 158 | 76 | 25 | 18 | |
| 2815 | 1033 | 327 | 125 | 40 | 17 | 8 |
| n\d | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|
| 8 | 0.4182 | 0.4123 | 0.4872 | 0.5 | 0.6 | |||
| 0.4097 | 0.3895 | 0.4688 | 0.3636 | 0.4 | ||||
| 9 | 1.2978 | 1.3333 | 1.4085 | 1.4074 | 1.5455 | 1.2 | ||
| 1.1837 | 1.2472 | 1.3519 | 1.1429 | 1.3333 | 1 | |||
| 10 | 2.7825 | 2.6995 | 2.9487 | 2.8214 | 3.4545 | 2.5 | 3.6 | |
| 2.3576 | 2.4654 | 2.7712 | 2.7778 | 2.5 | 2.125 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, H.; Zhou, S.; Yan, W.; Wang, S. Study on DNA Storage Encoding Based IAOA under Innovation Constraints. Curr. Issues Mol. Biol. 2023, 45, 3573-3590. https://doi.org/10.3390/cimb45040233
Du H, Zhou S, Yan W, Wang S. Study on DNA Storage Encoding Based IAOA under Innovation Constraints. Current Issues in Molecular Biology. 2023; 45(4):3573-3590. https://doi.org/10.3390/cimb45040233
Chicago/Turabian StyleDu, Haigui, Shihua Zhou, WeiQi Yan, and Sijie Wang. 2023. "Study on DNA Storage Encoding Based IAOA under Innovation Constraints" Current Issues in Molecular Biology 45, no. 4: 3573-3590. https://doi.org/10.3390/cimb45040233
APA StyleDu, H., Zhou, S., Yan, W., & Wang, S. (2023). Study on DNA Storage Encoding Based IAOA under Innovation Constraints. Current Issues in Molecular Biology, 45(4), 3573-3590. https://doi.org/10.3390/cimb45040233