
DDIT4 Downregulation by siRNA Approach Increases the Activity of Proteins Regulating Fatty Acid Metabolism upon Aspirin Treatment in Human Breast Cancer Cells

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture Conditions, and Treatment
2.2. Chemicals and Antibodies
2.3. Western Blot Analysis
2.4. Cell Transfection
2.5. Comparison of Protein Fold Change in Transfected Cells
2.6. Statistical Analysis
3. Results
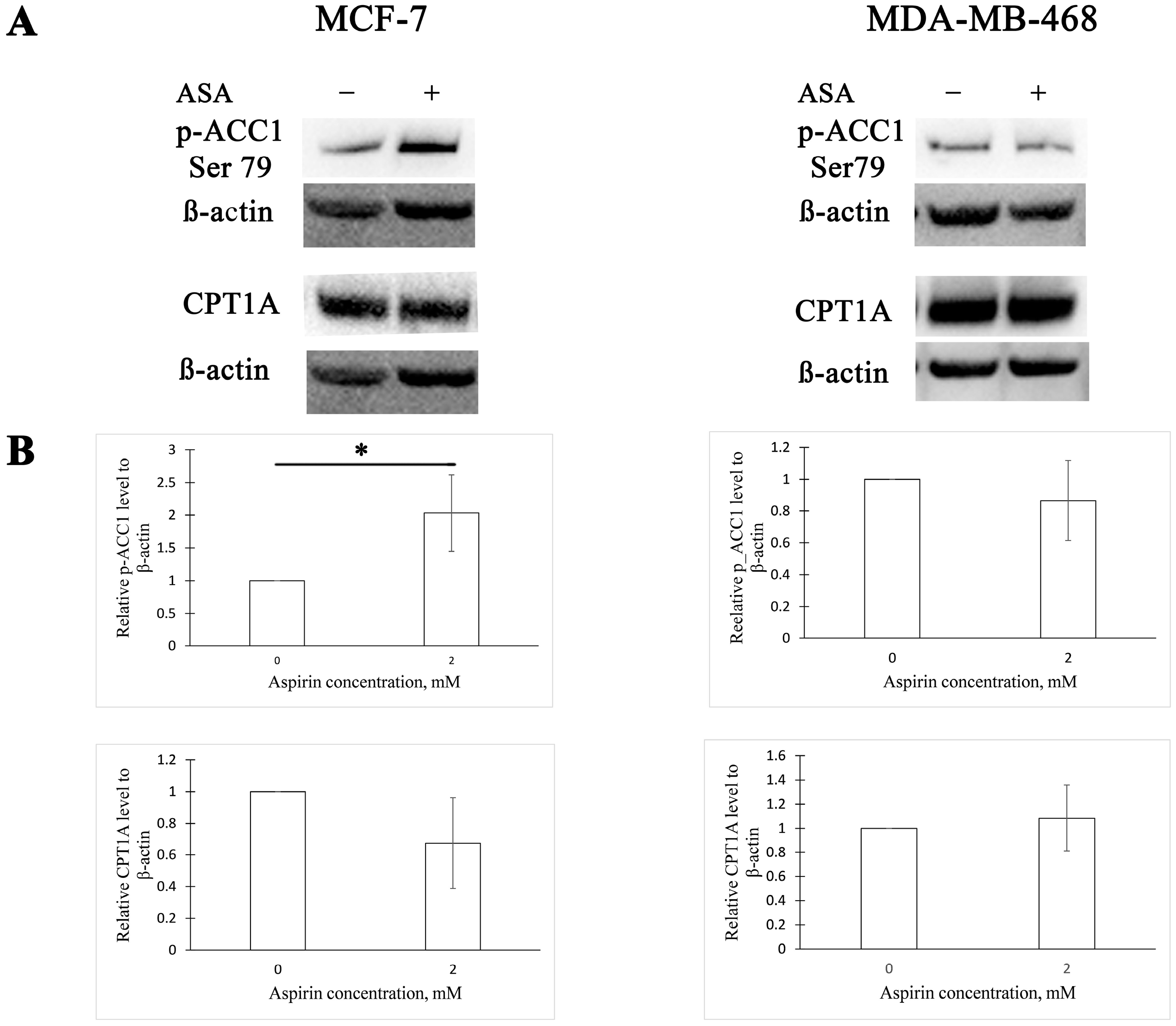
3.1. Aspirin Inhibits ACC1 Activity in MCF-7 Cells
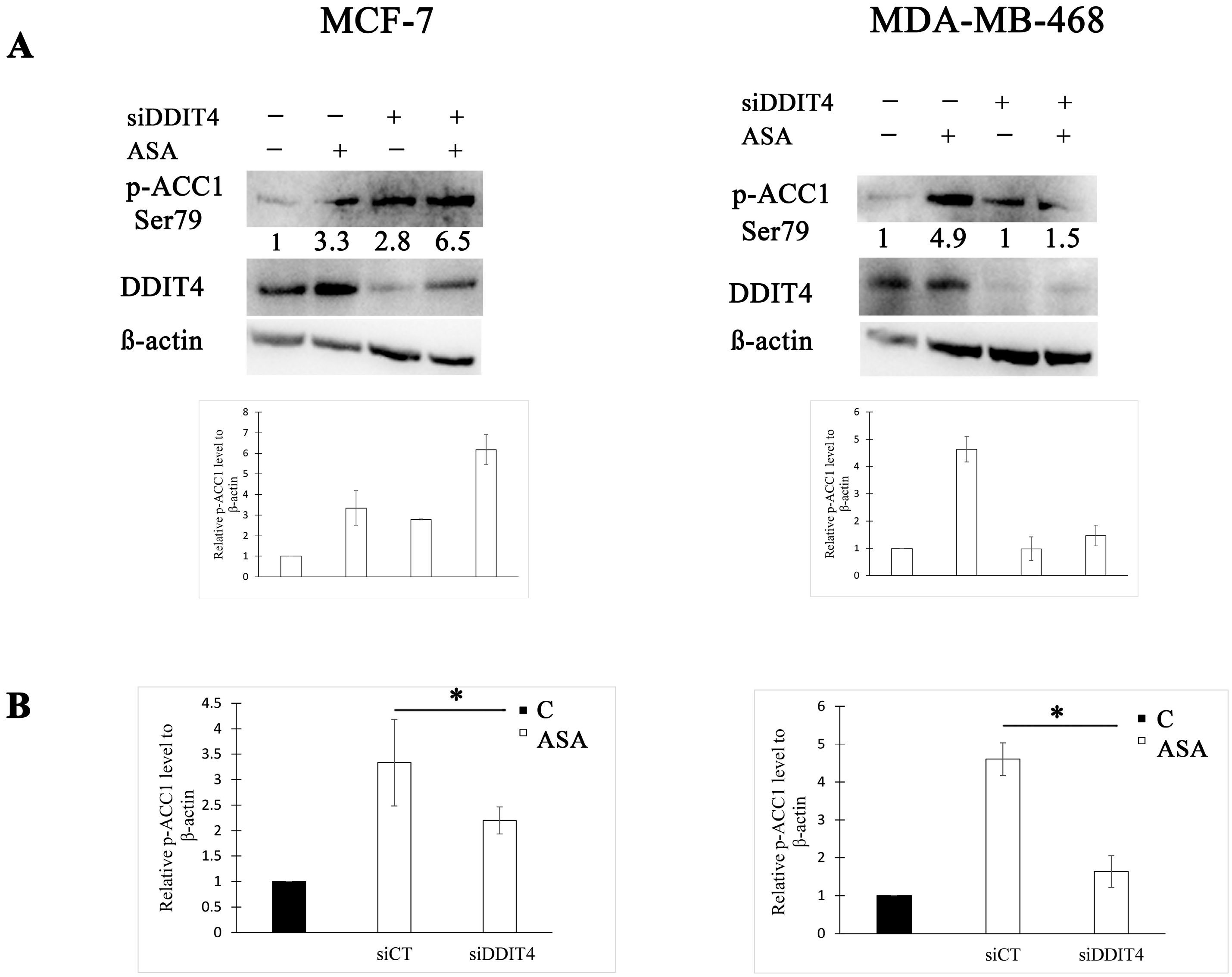
3.2. DDIT4 Knockdown Enhances ACC1 Activity following Aspirin Treatment in BC Cell Lines
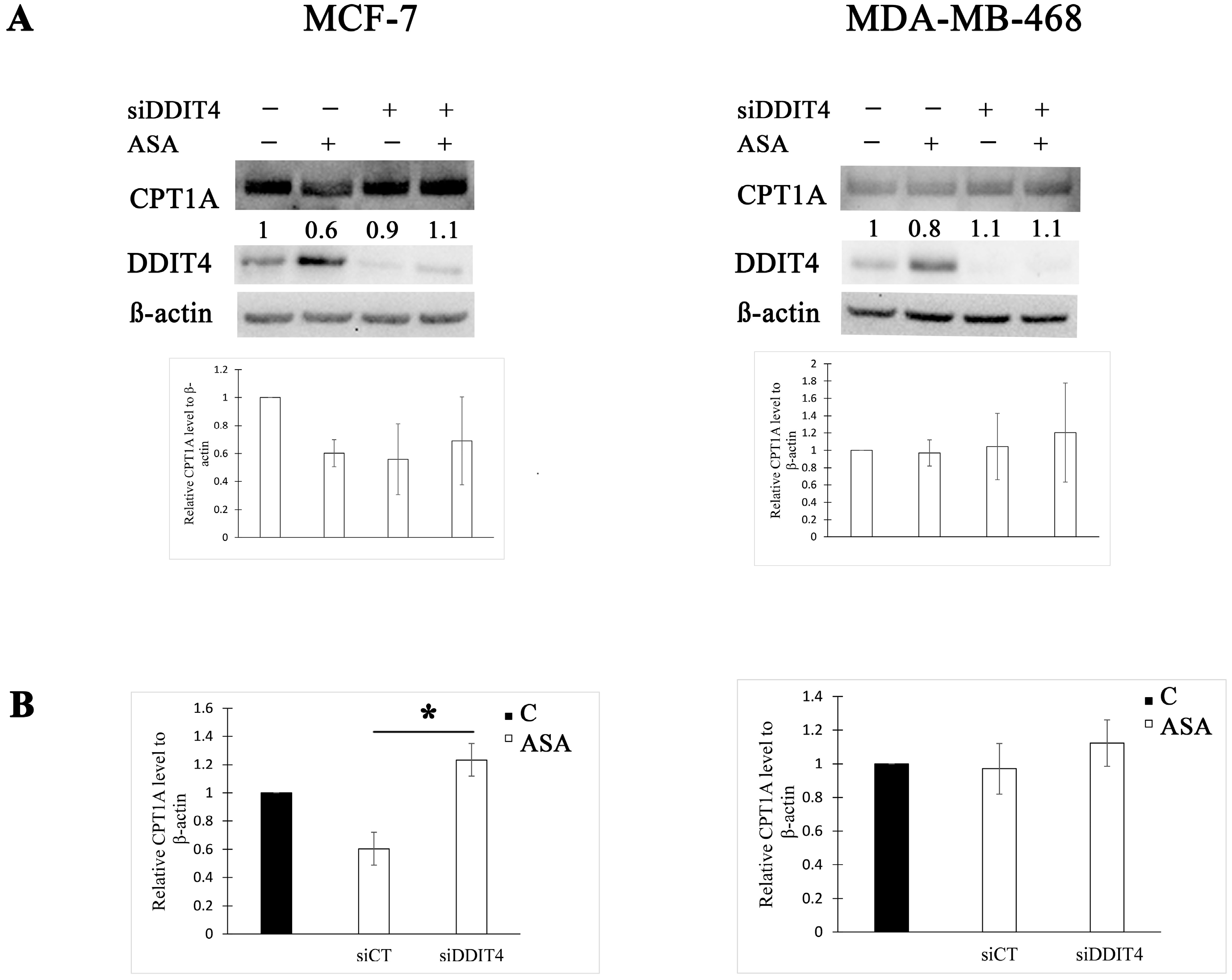
3.3. DDIT4 Knockdown Increases CPT1 Expression after Aspirin Treatment in MCF-7 Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tatham, M.H.; Cole, C.; Scullion, P.; Wilkie, R.; Westwood, N.J.; Stark, L.A.; Hay, R.T. A proteomic approach to analyze the aspirin-mediated lysine acetylome. Mol. Cell. Proteom. 2017, 16, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Rosato, V.; Gallus, S.; Cuzick, J.; La Vecchia, C. Aspirin and cancer risk: A quantitative review to 2011. Ann. Oncol. 2012, 23, 1403–1415. [Google Scholar] [CrossRef]
- Zhong, S.; Zhang, X.; Chen, L.; Ma, T.; Tang, J.; Zhao, J. Association between aspirin use and mortality in breast cancer patients: A meta-analysis of observational studies. Breast Cancer Res. Treat. 2015, 150, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Jamieson, G.G.; Wu, T.C.; Zhu, G.J.; Drew, P.A. A preliminary study on the postoperative survival of patients given aspirin after resection for squamous cell carcinoma of the esophagus or adenocarcinoma of the cardia. Ann. Surg. Oncol. 2009, 16, 1397–1402. [Google Scholar] [CrossRef]
- Van Staalduinen, J.; Frouws, M.; Reimers, M.; Bastiaannet, E.; van Herk-Sukel, M.P.; Lemmens, V.; de Steur, W.O.; Hartgrink, H.H.; van de Velde, C.J.; Liefers, G.J. The effect of aspirin and nonsteroidal anti-inflammatory drug use after diagnosis on survival of oesophageal cancer patients. Br. J. Cancer. 2016, 114, 1053–1059. [Google Scholar] [CrossRef]
- Zaorsky, N.G.; Buyyounouski, M.K.; Li, T.; Horwitz, E.M. Aspirin and statin nonuse associated with early biochemical failure after prostate radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, e13–e17. [Google Scholar] [CrossRef]
- Choe, K.S.; Cowan, J.E.; Chan, J.M.; Carroll, P.R.; D’Amico, A.V.; Liauw, S.L. Aspirin use and the risk of prostate cancer mortality in men treated with prostatectomy or radiotherapy. J. Clin. Oncol. 2012, 30, 3540–3544. [Google Scholar] [CrossRef]
- Jacobs, E.J.; Newton, C.C.; Stevens, V.L.; Campbell, P.T.; Freedland, S.J.; Gapstur, S.M. Daily aspirin use and prostate cancer–specific mortality in a large cohort of men with nonmetastatic prostate cancer. J. Clin. Oncol. 2014, 32, 3716–3722. [Google Scholar] [CrossRef]
- Holmes, M.D.; Chen, W.Y.; Li, L.; Hertzmark, E.; Spiegelman, D.; Hankinson, S.E. Aspirin intake and survival after breast cancer. J. Clin. Oncol. 2010, 28, 1467. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.M.; Sullivan, F.M.; Thompson, A.M.; McCowan, C. Aspirin use and survival after the diagnosis of breast cancer: A population-based cohort study. Br. J. Cancer 2014, 111, 623–627. [Google Scholar] [CrossRef]
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin use and survival after diagnosis of colorectal cancer. JAMA 2009, 302, 649–658. [Google Scholar] [CrossRef]
- Bastiaannet, E.; Sampieri, K.; Dekkers, O.M.; De Craen, A.J.; van Herk-Sukel, M.P.; Lemmens, V.; Van Den Broek, C.B.; Coebergh, J.W.; Herings, R.M.; Van De Velde, C.J.; et al. Use of aspirin postdiagnosis improves survival for colon cancer patients. Br. J. Cancer 2012, 106, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- McCowan, C.; Munro, A.J.; Donnan, P.T.; Steele, R.J. Use of aspirin post-diagnosis in a cohort of patients with colorectal cancer and its association with all-cause and colorectal cancer specific mortality. Eur. J. Cancer 2013, 49, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.J.; Grainge, M.J.; Card, T.R. Aspirin and other non-steroidal anti-inflammatory drug use and colorectal cancer survival: A cohort study. Br. J. Cancer 2012, 107, 1602–1607. [Google Scholar] [CrossRef] [PubMed]
- Bains, S.; Mahic, M.; Cvancarova, M.; Yaqub, S.; Dørum, L.M.; Bjørnbeth, B.A.; Møller, B.; Brudvik, K.W.; Tasken, K. Impact of aspirin as secondary prevention in an unselected cohort of 25,644 patients with colorectal cancer: A population-based study. J. Clin. Oncol. 2015, 33, 3504. [Google Scholar] [CrossRef]
- Ng, K.; Meyerhardt, J.A.; Chan, A.T.; Sato, K.; Chan, J.A.; Niedzwiecki, D.; Saltz, L.B.; Mayer, R.J.; Benson, A.B., III; Schaefer, P.L.; et al. Aspirin and COX-2 inhibitor use in patients with stage III colon cancer. J. Natl. Cancer Inst. 2015, 107, dju345. [Google Scholar] [CrossRef]
- Bashir, A.U.; Kankipati, C.S.; Jones, S.; Newman, R.M.; Safrany, S.T.; Perry, C.J.; Nicholl, I.D. A novel mechanism for the anticancer activity of aspirin and salicylates. Int. J. Oncol. 2019, 54, 1256–1270. [Google Scholar] [CrossRef]
- Heer, E.; Harper, A.; Escandor, N.; Sung, H.; McCormack, V.; Fidler-Benaoudia, M.M. Global burden and trends in premenopausal and postmenopausal breast cancer: A population-based study. Lancet Glob. Health 2020, 8, e1027–e1037. [Google Scholar] [CrossRef]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef]
- Foulon, A.; Theret, P.; Rodat-Despoix, L.; Kischel, P. Beyond chemotherapies: Recent strategies in breast cancer treatment. Cancers 2020, 12, 2634. [Google Scholar] [CrossRef]
- Grancher, A.; Michel, P.; Di Fiore, F.; Sefrioui, D. Colorectal cancer chemoprevention: Is aspirin still in the game? Cancer Biol. Ther. 2022, 23, 446–461. [Google Scholar] [CrossRef]
- Ward, A.V.; Anderson, S.M.; Sartorius, C.A. Advances in analyzing the breast cancer lipidome and its relevance to disease progression and treatment. J. Mammary Gland Biol. Neoplasia 2021, 26, 399–417. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated fat oxidation, mechanisms, and therapeutic potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Cebrián, N.; Domingo-Ortí, I.; Poveda, J.L.; Vicent, M.J.; Puchades-Carrasco, L.; Pineda-Lucena, A. Multi-omic approaches to breast cancer metabolic phenotyping: Applications in diagnosis, prognosis, and the development of novel treatments. Cancers 2021, 13, 4544. [Google Scholar] [CrossRef]
- Bao, J.; Zhu, L.; Zhu, Q.; Su, J.; Liu, M.; Huang, W. SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol. Lett. 2016, 12, 2409–2416. [Google Scholar] [CrossRef] [PubMed]
- Sebestyén, A.; Dankó, T.; Sztankovics, D.; Moldvai, D.; Raffay, R.; Cervi, C.; Krencz, I.; Zsiros, V.; Jeney, A.; Petővári, G. The role of metabolic ecosystem in cancer progression—Metabolic plasticity and mTOR hyperactivity in tumor tissues. Cancer Metastasis Rev. 2021, 40, 989–1033. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.A.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The carnitine system and cancer metabolic plasticity. Cell Death Dis. 2018, 9, 228. [Google Scholar] [CrossRef]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef]
- Hao, P.; Yu, J.; Ward, R.; Liu, Y.; Hao, Q.; An, S.; Xu, T. Eukaryotic translation initiation factors as promising targets in cancer therapy. Cell Commun. Signal. 2020, 18, 175. [Google Scholar] [CrossRef]
- Zhu, Y.; Lin, X.; Zhou, X.; Prochownik, E.V.; Wang, F.; Li, Y. Posttranslational control of lipogenesis in the tumor microenvironment. J. Hematol. Oncol. 2022, 15, 120. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, W.; Li, S.; Guo, D.; He, J.; Wang, Y. Acetyl-CoA carboxylases and diseases. Front. Oncol. 2022, 12, 836058. [Google Scholar] [CrossRef] [PubMed]
- Koobotse, M.; Holly, J.; Perks, C. Elucidating the novel BRCA1 function as a non-genomic metabolic restraint in ER-positive breast cancer cell lines. Oncotarget 2018, 9, 33562. [Google Scholar] [CrossRef]
- Chajes, V.; Cambot, M.; Moreau, K.; Lenoir, G.M.; Joulin, V. Acetyl-CoA carboxylase α is essential to breast cancer cell survival. Cancer Res. 2006, 66, 5287–5294. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.E. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487. [Google Scholar] [CrossRef]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- Das, M.; Giannoudis, A.; Sharma, V. The role of CPT1A as a biomarker of breast cancer progression: A bioinformatic approach. Sci. Rep. 2022, 12, 16441. [Google Scholar] [CrossRef]
- Gatza, M.L.; Silva, G.O.; Parker, J.S.; Fan, C.; Perou, C.M. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat. Genet. 2014, 46, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.D.; Kahle, K.T.; Sun, T.; Meerbrey, K.L.; Schlabach, M.R.; Schmitt, E.M.; Skinner, S.O.; Xu, Q.; Li, M.Z.; Hartman, Z.C.; et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 2012, 335, 348–353. [Google Scholar] [CrossRef]
- Tan, Z.; Zou, Y.; Zhu, M.; Luo, Z.; Wu, T.; Zheng, C.; Xie, A.; Wang, H.; Fang, S.; Liu, S.; et al. Carnitine palmitoyl transferase 1A is a novel diagnostic and predictive biomarker for breast cancer. BMC Cancer 2021, 21, 409. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- Savukaitytė, A.; Gudoitytė, G.; Bartnykaitė, A.; Ugenskienė, R.; Juozaitytė, E. siRNA knockdown of REDD1 facilitates aspirin-mediated dephosphorylation of mTORC1 target 4E-BP1 in MDA-MB-468 human breast cancer cell line. Cancer Manag. Res. 2021, 13, 1123–1133. [Google Scholar] [CrossRef]
- Dovizio, M.; Tacconelli, S.; Sostres, C.; Ricciotti, E.; Patrignani, P. Mechanistic and pharmacological issues of aspirin as an anticancer agent. Pharmaceuticals 2012, 5, 1346–1371. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Peng, Y.; Duan, W.; Tian, Y.; Zhang, J.; Hu, T.; Cai, Y.; Feng, Y.; Li, G. Aspirin regulates hepatocellular lipid metabolism by activating AMPK signaling pathway. J. Toxicol. Sci. 2015, 40, 127–136. [Google Scholar] [CrossRef]
- Henry, W.S.; Laszewski, T.; Tsang, T.; Beca, F.; Beck, A.H.; McAllister, S.S.; Toker, A. Aspirin Suppresses Growth in PI3K-Mutant Breast Cancer by Activating AMPK and Inhibiting mTORC1 SignalingAspirin and PI3K in Breast Cancer. Cancer Res. 2017, 77, 790–801. [Google Scholar] [CrossRef]
- Wu, Y.; Yan, B.; Xu, W.; Guo, L.; Wang, Z.; Li, G.; Hou, N.; Zhang, J.; Ling, R. Compound C enhances the anticancer effect of aspirin in HER-2-positive breast cancer by regulating lipid metabolism in an AMPK-independent pathway. Int. J. Biol. Sci. 2020, 16, 583. [Google Scholar] [CrossRef]
- Uppala, R.; Dudiak, B.; Beck, M.E.; Bharathi, S.S.; Zhang, Y.; Stolz, D.B.; Goetzman, E.S. Aspirin increases mitochondrial fatty acid oxidation. Biochem. Biophys. Res. Commun. 2017, 482, 346–351. [Google Scholar] [CrossRef]
- Di Minno, A.; Porro, B.; Turnu, L.; Manega, C.M.; Eligini, S.; Barbieri, S.; Chiesa, M.; Poggio, P.; Squellerio, I.; Anesi, A.; et al. Untargeted metabolomics to go beyond the canonical effect of acetylsalicylic acid. J. Clin. Med. 2019, 9, 51. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.A.; Rolfo, C.; Raez, L.E.; Prado, A.; Araujo, J.M.; Bravo, L.; Fajardo, W.; Morante, Z.D.; Aguilar, A.; Neciosup, S.P.; et al. In silico evaluation of DNA Damage Inducible Transcript 4 gene (DDIT4) as prognostic biomarker in several malignancies. Sci. Rep. 2017, 7, 1526. [Google Scholar] [CrossRef] [PubMed]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2–mTOR signaling and tumor suppression through REDD1-mediated 14–3–3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savukaitytė, A.; Bartnykaitė, A.; Bekampytė, J.; Ugenskienė, R.; Juozaitytė, E. DDIT4 Downregulation by siRNA Approach Increases the Activity of Proteins Regulating Fatty Acid Metabolism upon Aspirin Treatment in Human Breast Cancer Cells. Curr. Issues Mol. Biol. 2023, 45, 4665-4674. https://doi.org/10.3390/cimb45060296
Savukaitytė A, Bartnykaitė A, Bekampytė J, Ugenskienė R, Juozaitytė E. DDIT4 Downregulation by siRNA Approach Increases the Activity of Proteins Regulating Fatty Acid Metabolism upon Aspirin Treatment in Human Breast Cancer Cells. Current Issues in Molecular Biology. 2023; 45(6):4665-4674. https://doi.org/10.3390/cimb45060296
Chicago/Turabian StyleSavukaitytė, Aistė, Agnė Bartnykaitė, Justina Bekampytė, Rasa Ugenskienė, and Elona Juozaitytė. 2023. "DDIT4 Downregulation by siRNA Approach Increases the Activity of Proteins Regulating Fatty Acid Metabolism upon Aspirin Treatment in Human Breast Cancer Cells" Current Issues in Molecular Biology 45, no. 6: 4665-4674. https://doi.org/10.3390/cimb45060296
APA StyleSavukaitytė, A., Bartnykaitė, A., Bekampytė, J., Ugenskienė, R., & Juozaitytė, E. (2023). DDIT4 Downregulation by siRNA Approach Increases the Activity of Proteins Regulating Fatty Acid Metabolism upon Aspirin Treatment in Human Breast Cancer Cells. Current Issues in Molecular Biology, 45(6), 4665-4674. https://doi.org/10.3390/cimb45060296