
Adenosine A2A Receptor Activation Regulates Niemann–Pick C1 Expression and Localization in Macrophages

 ,
,  ,
,  ,
,
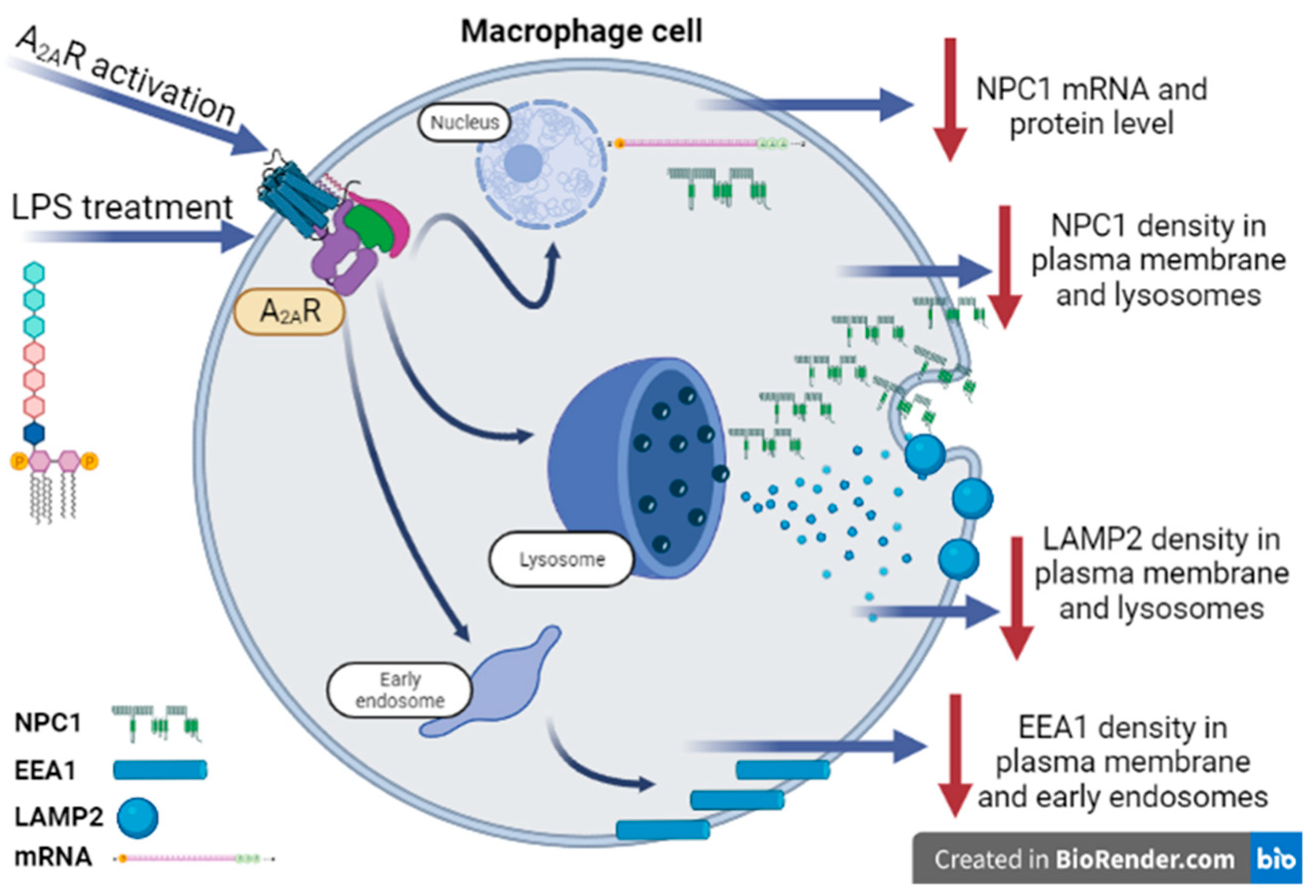
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Isolation of Mouse Peritoneal Macrophages
2.3. Animal Models
2.4. Cell Culture
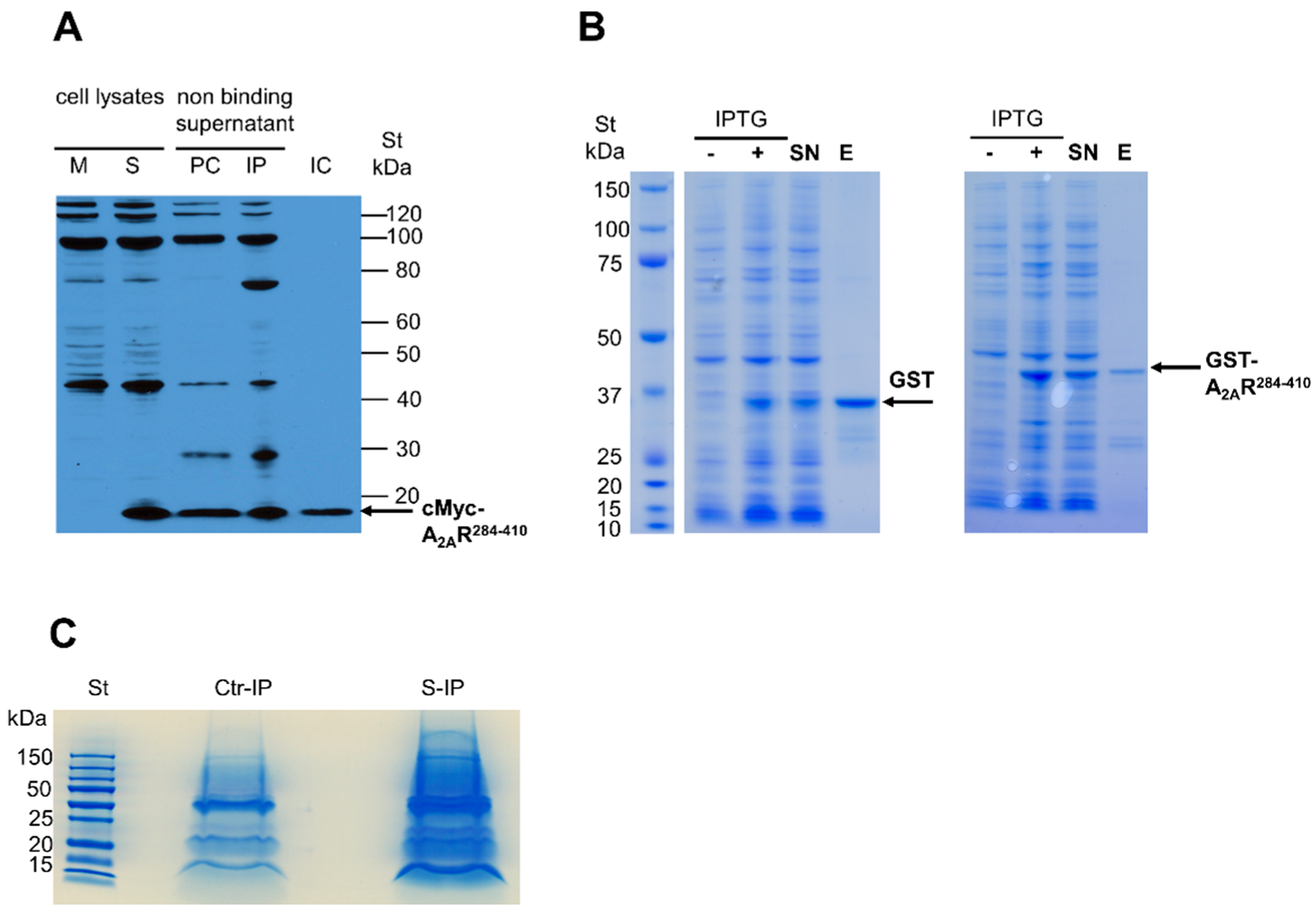
2.5. Immunoprecipitation of cMyc-A2AR284–410
2.6. Purification of GST-A2AR284–410 Specific Pull-Down Complex
2.7. Mass Spectrometry
2.8. Pharmacological Treatment of Macrophages
2.9. Protein Isolation
2.10. RNA Extraction and q-RT-PCR
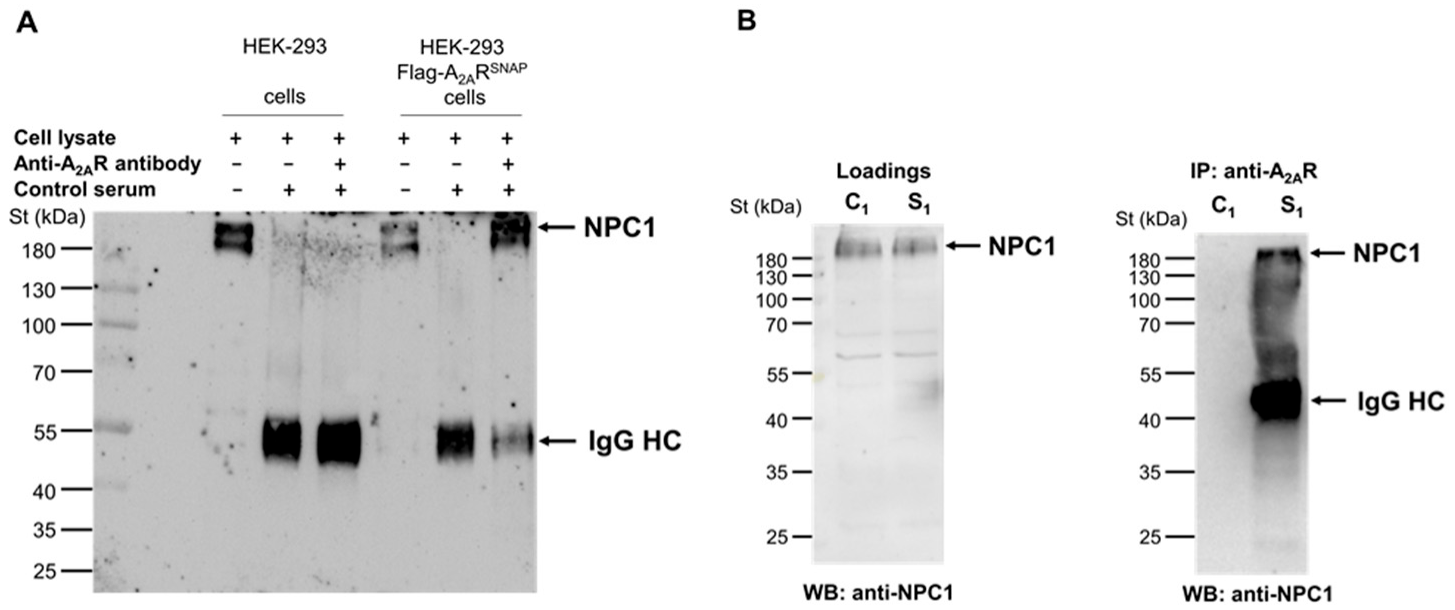
2.11. Immunoprecipitation of cMyc-A2AR284–410 and NPC1 Proteins
2.12. Immunoblot
2.13. Immunostaining of LAMP2, EEA1 and NPC1 Protein
2.14. Laser Scanning Cytometry
2.15. Statistical Analyses
3. Results
3.1. Identification of A2AR-Interacting Proteins by Co-Immunoprecipitation and Pull-Down Experiments Coupled with Mass Spectrometry Detection
3.2. Validation of A2AR and Endogenous NPC1 Interaction in Living Cells
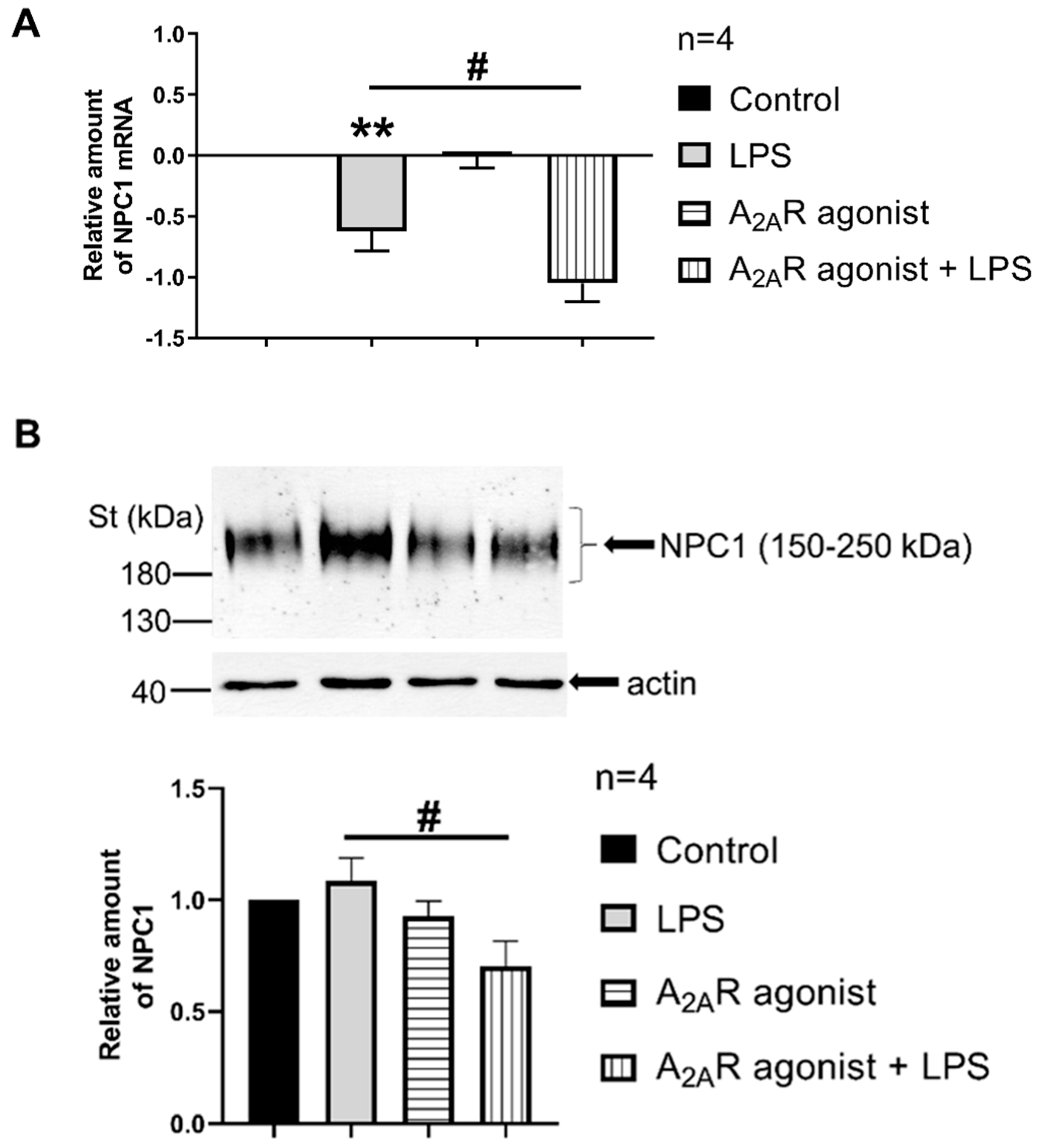
3.3. A2AR Activation Reduces NPC1 mRNA Expression and Protein Density in LPS-Activated Macrophages
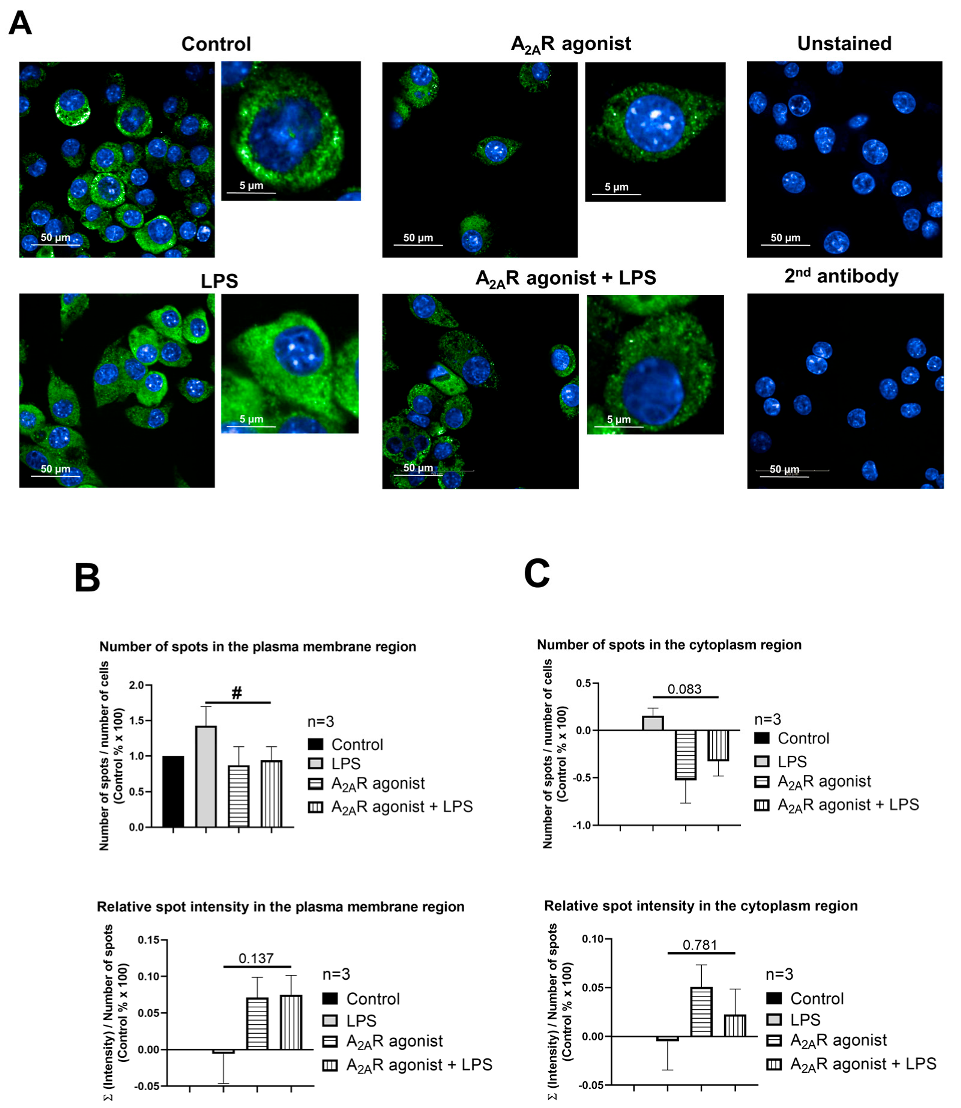
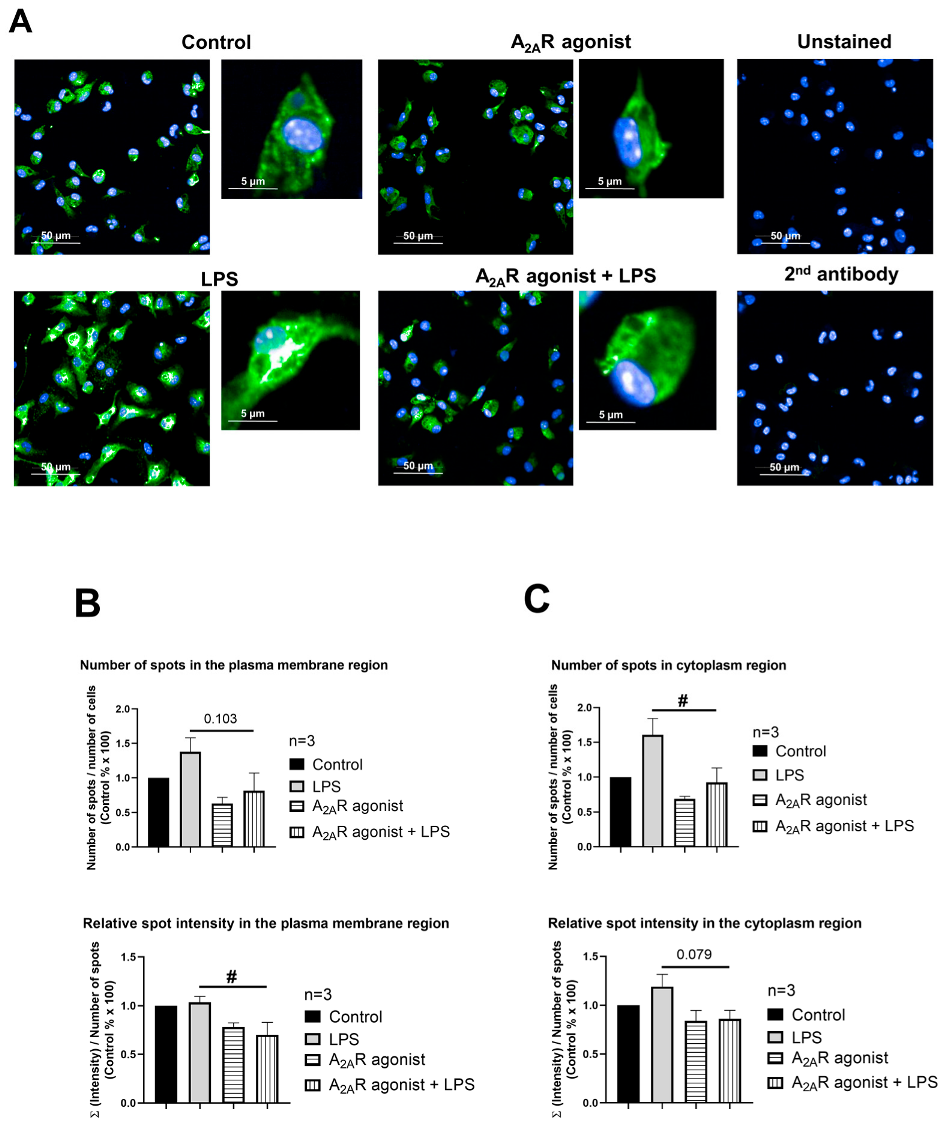
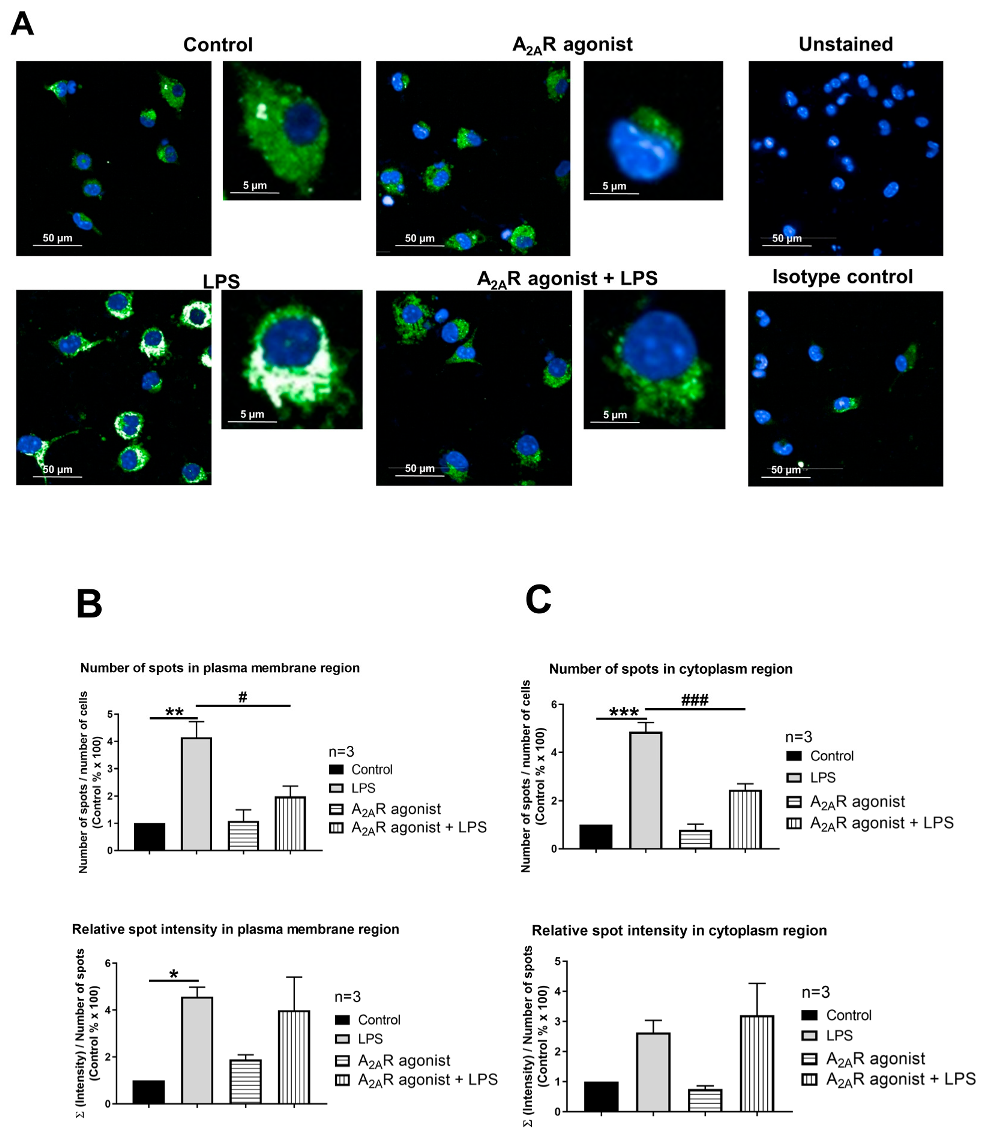
3.4. NPC1 Cell Surface Expression in Mouse Macrophages Is Controlled by A2AR Activation
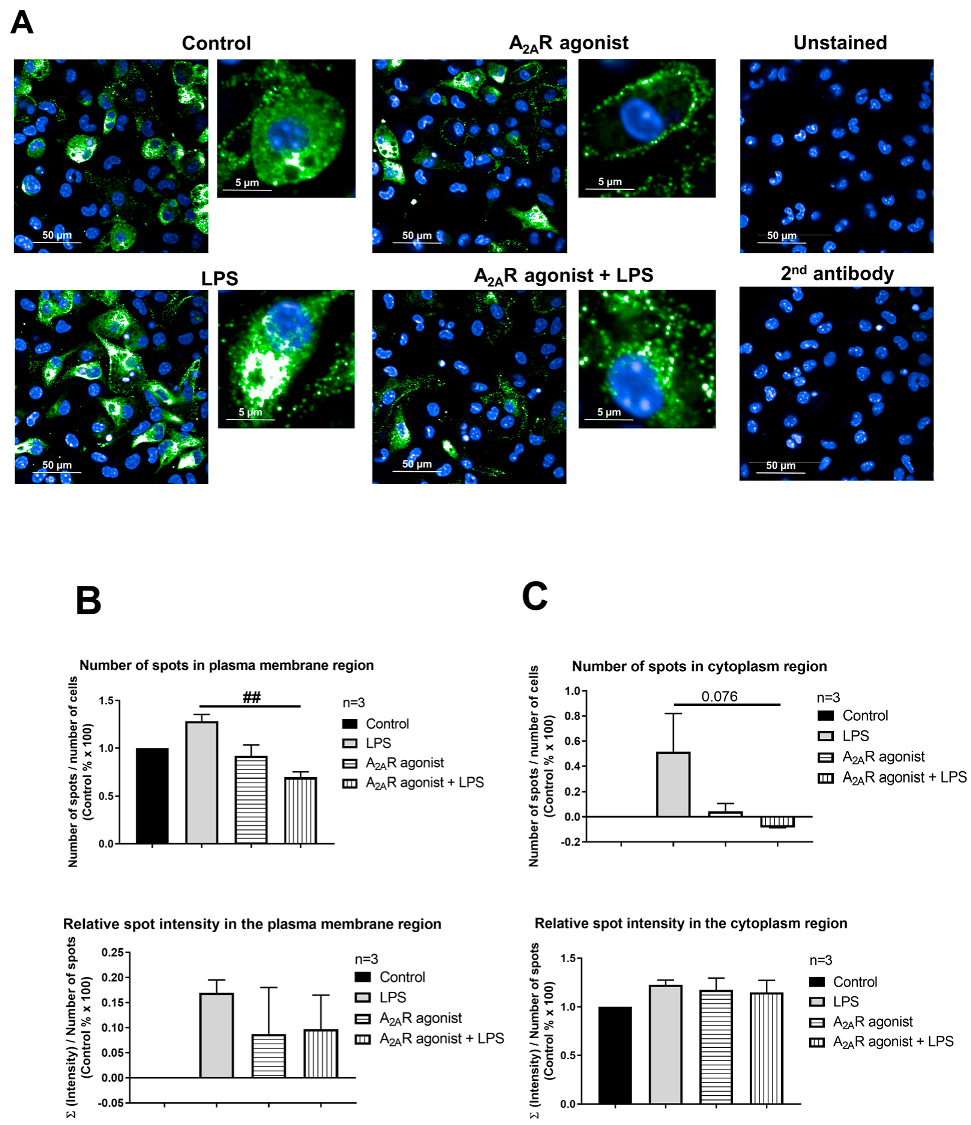
3.5. A2AR Stimulation Decreases Lysosomal-Associated Membrane Protein 2 (LAMP2) Expression in Mouse Macrophages
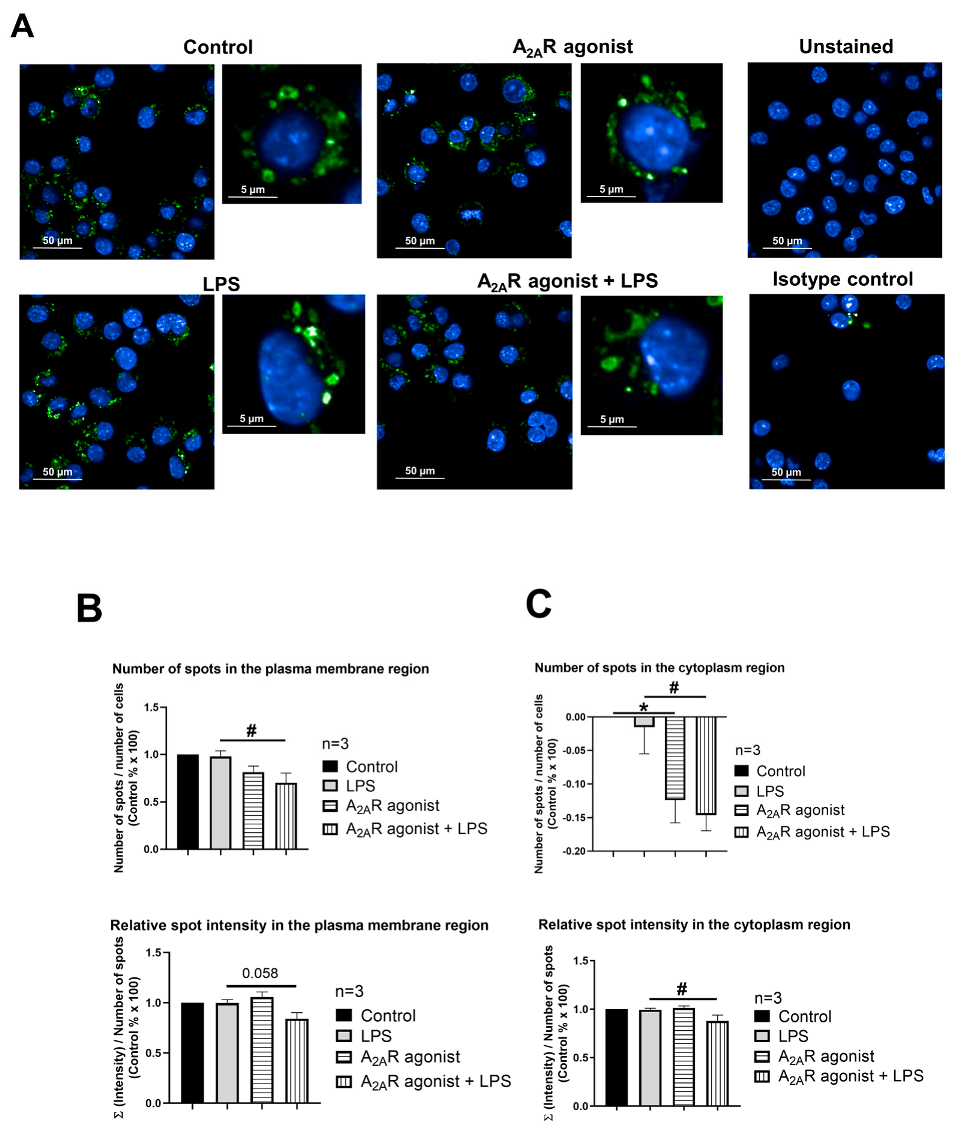
3.6. A2AR Activation Modulates Early Endosome Antigen 1 (EEA1) Expression in Mouse Macrophages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olah, M.E.; Stiles, G.L. Adenosine receptors. Annu. Rev. Physiol. 1992, 54, 211–225. [Google Scholar] [CrossRef] [PubMed]
- IJzerman, A.P.; Jacobson, K.A.; Muller, C.E.; Cronstein, B.N.; Cunha, R.A. International Union of Basic and Clinical Pharmacology. CXII: Adenosine Receptors: A Further Update. Pharmacol. Rev. 2022, 74, 340–372. [Google Scholar] [CrossRef]
- Ohta, A.; Sitkovsky, M. The adenosinergic immunomodulatory drugs. Curr. Opin. Pharmacol. 2009, 9, 501–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasko, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef]
- Ferre, S.; Ciruela, F.; Dessauer, C.W.; Gonzalez-Maeso, J.; Hebert, T.E.; Jockers, R.; Logothetis, D.E.; Pardo, L. G protein-coupled receptor-effector macromolecular membrane assemblies (GEMMAs). Pharmacol. Ther. 2022, 231, 107977. [Google Scholar] [CrossRef]
- Ciruela, F.; Albergaria, C.; Soriano, A.; Cuffi, L.; Carbonell, L.; Sanchez, S.; Gandia, J.; Fernandez-Duenas, V. Adenosine receptors interacting proteins (ARIPs): Behind the biology of adenosine signaling. Biochim. Biophys. Acta 2010, 1798, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Keuerleber, S.; Gsandtner, I.; Freissmuth, M. From cradle to twilight: The carboxyl terminus directs the fate of the A(2A)-adenosine receptor. Biochim. Biophys. Acta 2011, 1808, 1350–1357. [Google Scholar] [CrossRef] [Green Version]
- Canals, M.; Marcellino, D.; Fanelli, F.; Ciruela, F.; de Benedetti, P.; Goldberg, S.R.; Neve, K.; Fuxe, K.; Agnati, L.F.; Woods, A.S.; et al. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: Qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J. Biol. Chem. 2003, 278, 46741–46749. [Google Scholar] [CrossRef] [Green Version]
- Fuxe, K.; Agnati, L.F.; Jacobsen, K.; Hillion, J.; Canals, M.; Torvinen, M.; Tinner-Staines, B.; Staines, W.; Rosin, D.; Terasmaa, A.; et al. Receptor heteromerization in adenosine A2A receptor signaling: Relevance for striatal function and Parkinson’s disease. Neurology 2003, 61, S19–S23. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Romero-Fernandez, W.; Tarakanov, A.O.; Gomez-Soler, M.; Corrales, F.; Marcellino, D.; Narvaez, M.; Frankowska, M.; Flajolet, M.; Heintz, N.; et al. Characterization of the A2AR-D2R interface: Focus on the role of the C-terminal tail and the transmembrane helices. Biochem. Biophys. Res. Commun. 2010, 402, 801–807. [Google Scholar] [CrossRef]
- Ferre, S.; Sarasola, L.I.; Quiroz, C.; Ciruela, F. Presynaptic adenosine receptor heteromers as key modulators of glutamatergic and dopaminergic neurotransmission in the striatum. Neuropharmacology 2023, 223, 109329. [Google Scholar] [CrossRef]
- Chen, J.F.; Cunha, R.A. The belated US FDA approval of the adenosine A(2A) receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef]
- Morato, X.; Lujan, R.; Lopez-Cano, M.; Gandia, J.; Stagljar, I.; Watanabe, M.; Cunha, R.A.; Fernandez-Duenas, V.; Ciruela, F. The Parkinson’s disease-associated GPR37 receptor interacts with striatal adenosine A(2A) receptor controlling its cell surface expression and function in vivo. Sci. Rep. 2017, 7, 9452. [Google Scholar] [CrossRef] [Green Version]
- Sokolina, K.; Kittanakom, S.; Snider, J.; Kotlyar, M.; Maurice, P.; Gandia, J.; Benleulmi-Chaachoua, A.; Tadagaki, K.; Oishi, A.; Wong, V.; et al. Systematic protein-protein interaction mapping for clinically relevant human GPCRs. Mol. Syst. Biol. 2017, 13, 918. [Google Scholar] [CrossRef]
- Scianni, M.; Antonilli, L.; Chece, G.; Cristalli, G.; Di Castro, M.A.; Limatola, C.; Maggi, L. Fractalkine (CX3CL1) enhances hippocampal N-methyl-D-aspartate receptor (NMDAR) function via D-serine and adenosine receptor type A2 (A2AR) activity. J. Neuroinflamm. 2013, 10, 876. [Google Scholar] [CrossRef] [Green Version]
- Constantino, L.C.; Pamplona, F.A.; Matheus, F.C.; de Carvalho, C.R.; Ludka, F.K.; Massari, C.M.; Boeck, C.R.; Prediger, R.D.; Tasca, C.I. Functional interplay between adenosine A(2A) receptor and NMDA preconditioning in fear memory and glutamate uptake in the mice hippocampus. Neurobiol. Learn. Mem. 2021, 180, 107422. [Google Scholar] [CrossRef]
- Yamada, K.; Kobayashi, M.; Kanda, T. Involvement of adenosine A2A receptors in depression and anxiety. Int. Rev. Neurobiol. 2014, 119, 373–393. [Google Scholar] [CrossRef]
- Krugel, U. Purinergic receptors in psychiatric disorders. Neuropharmacology 2016, 104, 212–225. [Google Scholar] [CrossRef]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Van der Jeugd, A.; et al. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry 2016, 21, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes, A.P.; Machado, N.J.; Goncalves, N.; Kaster, M.P.; Simoes, A.T.; Nunes, A.; Pereira de Almeida, L.; Goosens, K.A.; Rial, D.; Cunha, R.A. Adenosine A(2A) Receptors in the Amygdala Control Synaptic Plasticity and Contextual Fear Memory. Neuropsychopharmacology 2016, 41, 2862–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skopal, A.; Keki, T.; Toth, P.A.; Csoka, B.; Koscso, B.; Nemeth, Z.H.; Antonioli, L.; Ivessa, A.; Ciruela, F.; Virag, L.; et al. Cathepsin D interacts with adenosine A(2A) receptors in mouse macrophages to modulate cell surface localization and inflammatory signaling. J. Biol. Chem. 2022, 298, 101888. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Kuhel, D.G.; Chen, J.F.; Schwarzschild, M.A.; Deitch, E.A.; Mabley, J.G.; Marton, A.; Szabo, C. Adenosine inhibits IL-12 and TNF-α production via adenosine A2a receptor-dependent and independent mechanisms. Faseb J. 2000, 14, 2065–2074. [Google Scholar] [CrossRef] [Green Version]
- Hasko, G.; Xu, D.Z.; Lu, Q.; Nemeth, Z.H.; Jabush, J.; Berezina, T.L.; Zaets, S.B.; Csoka, B.; Deitch, E.A. Adenosine A2A receptor activation reduces lung injury in trauma/hemorrhagic shock. Crit. Care Med. 2006, 34, 1119–1125. [Google Scholar] [CrossRef]
- Nemeth, Z.H.; Csoka, B.; Wilmanski, J.; Xu, D.; Lu, Q.; Ledent, C.; Deitch, E.A.; Pacher, P.; Spolarics, Z.; Hasko, G. Adenosine A2A receptor inactivation increases survival in polymicrobial sepsis. J. Immunol. 2006, 176, 5616–5626. [Google Scholar] [CrossRef] [Green Version]
- Csoka, B.; Nemeth, Z.H.; Virag, L.; Gergely, P.; Leibovich, S.J.; Pacher, P.; Sun, C.X.; Blackburn, M.R.; Vizi, E.S.; Deitch, E.A.; et al. A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood 2007, 110, 2685–2695. [Google Scholar] [CrossRef] [Green Version]
- Csoka, B.; Himer, L.; Selmeczy, Z.; Vizi, E.S.; Pacher, P.; Ledent, C.; Deitch, E.A.; Spolarics, Z.; Nemeth, Z.H.; Hasko, G. Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J. 2008, 22, 3491–3499. [Google Scholar] [CrossRef] [Green Version]
- Hasko, G.; Szabo, C.; Nemeth, Z.H.; Kvetan, V.; Pastores, S.M.; Vizi, E.S. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J. Immunol. 1996, 157, 4634–4640. [Google Scholar] [CrossRef]
- Ingwersen, J.; Wingerath, B.; Graf, J.; Lepka, K.; Hofrichter, M.; Schroter, F.; Wedekind, F.; Bauer, A.; Schrader, J.; Hartung, H.P.; et al. Dual roles of the adenosine A2a receptor in autoimmune neuroinflammation. J. Neuroinflamm. 2016, 13, 48. [Google Scholar] [CrossRef] [Green Version]
- Mazzon, E.; Esposito, E.; Impellizzeri, D.; Di Paola, R.; Melani, A.; Bramanti, P.; Pedata, F.; Cuzzocrea, S. CGS 21680, an agonist of the adenosine (A2A) receptor, reduces progression of murine type II collagen-induced arthritis. J. Rheumatol. 2011, 38, 2119–2129. [Google Scholar] [CrossRef]
- Friebe, D.; Yang, T.; Schmidt, T.; Borg, N.; Steckel, B.; Ding, Z.; Schrader, J. Purinergic signaling on leukocytes infiltrating the LPS-injured lung. PLoS ONE 2014, 9, e95382. [Google Scholar] [CrossRef] [Green Version]
- Day, Y.J.; Huang, L.; McDuffie, M.J.; Rosin, D.L.; Ye, H.; Chen, J.F.; Schwarzschild, M.A.; Fink, J.S.; Linden, J.; Okusa, M.D. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J. Clin. Investig. 2003, 112, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Vitzthum, H.; Weiss, B.; Bachleitner, W.; Kramer, B.K.; Kurtz, A. Gene expression of adenosine receptors along the nephron. Kidney Int. 2004, 65, 1180–1190. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Fornai, M.; Colucci, R.; Ghisu, N.; Tuccori, M.; Del Tacca, M.; Blandizzi, C. Regulation of enteric functions by adenosine: Pathophysiological and pharmacological implications. Pharmacol. Ther. 2008, 120, 233–253. [Google Scholar] [CrossRef]
- Ferrante, A.; De Nuccio, C.; Pepponi, R.; Visentin, S.; Martire, A.; Bernardo, A.; Minghetti, L.; Popoli, P. Stimulation of adenosine A2A receptors reduces intracellular cholesterol accumulation and rescues mitochondrial abnormalities in human neural cell models of Niemann-Pick C1. Neuropharmacology 2016, 103, 155–162. [Google Scholar] [CrossRef]
- Visentin, S.; De Nuccio, C.; Bernardo, A.; Pepponi, R.; Ferrante, A.; Minghetti, L.; Popoli, P. The stimulation of adenosine A2A receptors ameliorates the pathological phenotype of fibroblasts from Niemann-Pick type C patients. J. Neurosci. 2013, 33, 15388–15393. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.R. NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem. 2019, 294, 1706–1709. [Google Scholar] [CrossRef] [Green Version]
- Umeda, A.; Fujita, H.; Kuronita, T.; Hirosako, K.; Himeno, M.; Tanaka, Y. Distribution and trafficking of MPR300 is normal in cells with cholesterol accumulated in late endocytic compartments: Evidence for early endosome-to-TGN trafficking of MPR300. J. Lipid Res. 2003, 44, 1821–1832. [Google Scholar] [CrossRef] [Green Version]
- Sümegi, M.; Hunyadi-Gulyás, É.; Medzihradszky, K.F.; Udvardy, A. 26S proteasome subunits are O-linked N-acetylglucosamine-modified in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2003, 312, 1284–1289. [Google Scholar] [CrossRef]
- Fernandez-Duenas, V.; Taura, J.J.; Cottet, M.; Gomez-Soler, M.; Lopez-Cano, M.; Ledent, C.; Watanabe, M.; Trinquet, E.; Pin, J.P.; Lujan, R.; et al. Untangling dopamine-adenosine receptor-receptor assembly in experimental parkinsonism in rats. Dis. Model Mech. 2015, 8, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, M.N.; Brown, M.S.; Seemann, J.; Goldstein, J.L.; Lu, F. Lysosomal cholesterol export reconstituted from fragments of Niemann-Pick C1. eLife 2018, 7, e38564. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Qin, K.; Li, N.; Han, C.; Cao, X. An endosomal LAPF is required for macrophage endocytosis and elimination of bacteria. Proc. Natl. Acad. Sci. USA 2019, 116, 12958–12963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominko, K.; Rastija, A.; Sobocanec, S.; Vidatic, L.; Meglaj, S.; Lovincic Babic, A.; Hutter-Paier, B.; Colombo, A.V.; Lichtenthaler, S.F.; Tahirovic, S.; et al. Impaired Retromer Function in Niemann-Pick Type C Disease Is Dependent on Intracellular Cholesterol Accumulation. Int. J. Mol. Sci. 2021, 22, 13256. [Google Scholar] [CrossRef]
- De Nuccio, C.; Bernardo, A.; Ferrante, A.; Pepponi, R.; Martire, A.; Falchi, M.; Visentin, S.; Popoli, P.; Minghetti, L. Adenosine A(2A) receptor stimulation restores cell functions and differentiation in Niemann-Pick type C-like oligodendrocytes. Sci. Rep. 2019, 9, 9782. [Google Scholar] [CrossRef] [Green Version]
- Filiberti, S.; Russo, M.; Lonardi, S.; Bugatti, M.; Vermi, W.; Tournier, C.; Giurisato, E. Self-Renewal of Macrophages: Tumor-Released Factors and Signaling Pathways. Biomedicines 2022, 10, 2709. [Google Scholar] [CrossRef]
- Huynh, K.K.; Gershenzon, E.; Grinstein, S. Cholesterol accumulation by macrophages impairs phagosome maturation. J. Biol. Chem. 2008, 283, 35745–35755. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Gomez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Tchikov, V.; Bertsch, U.; Fritsch, J.; Edelmann, B.; Schutze, S. Subcellular compartmentalization of TNF receptor-1 and CD95 signaling pathways. Eur. J. Cell Biol. 2011, 90, 467–475. [Google Scholar] [CrossRef]
- Bernardo, A.; De Nuccio, C.; Visentin, S.; Martire, A.; Minghetti, L.; Popoli, P.; Ferrante, A. Myelin Defects in Niemann-Pick Type C Disease: Mechanisms and Possible Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22, 8858. [Google Scholar] [CrossRef]
- Watari, H.; Blanchette-Mackie, E.J.; Dwyer, N.K.; Watari, M.; Burd, C.G.; Patel, S.; Pentchev, P.G.; Strauss, J.F., 3rd. Determinants of NPC1 expression and action: Key promoter regions, posttranscriptional control, and the importance of a “cysteine-rich” loop. Exp. Cell Res. 2000, 259, 247–256. [Google Scholar] [CrossRef]
- Carini, R.; Castino, R.; De Cesaris, M.G.; Splendore, R.; Demoz, M.; Albano, E.; Isidoro, C. Preconditioning-induced cytoprotection in hepatocytes requires Ca2+-dependent exocytosis of lysosomes. J. Cell Sci. 2004, 117, 1065–1077. [Google Scholar] [CrossRef] [Green Version]
- Min, B.K.; Suk, K.; Lee, W.H. Stimulation of CD107 affects LPS-induced cytokine secretion and cellular adhesion through the ERK signaling pathway in the human macrophage-like cell line, THP-1. Cell. Immunol. 2013, 281, 122–128. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward | Reverse |
|---|---|---|
| β2M | 5′-AGTATACTCACGCCACCCAC-3′ | 5′-CATGTCTCGATCCCAGTAGACG-3′ |
| NPC1 GAPDH | 5′-TTTGGTATGGAGAGTGTGGA-3′ 5′-ACAGTCCATGCCATCACTG-3′ | 5′-ACAGCAGAGACTGACATTGT-3′ 5′-GCCTGCTTCACCACCTTCTT-3′ |
| Antibody | Methods | Applied Concentration | Catalog Number; Supplier |
|---|---|---|---|
| anti-A2AR | IP | 4.25 µg/mL | AAR-002; Alomone labs (Jerusalem, Israel) |
| anti-cMyc | WB IP | 1.67 µg/mL 16.5 µg/mL | M5546, Sigma-Aldrich, (Budapest, Hungary) |
| anti-EEA1 | IS | 1 µg/mL | SAB4300682; Sigma Aldrich (Budapest, Hungary) |
| anti-NPC1 | WB IS | 1 µg/mL 5 µg/mL | NB400-148, Novus Biologicals (Centennial, CO, USA) |
| anti-rabbit-HRP | WB | 0.2 µg/mL | 7074S, Cell Signaling Technology (Danvers, MA, USA) |
| anti-Mouse-HRP | WB | 0.2 µg/mL | 7076S; Cell Signaling Technology (Danvers, MA, USA) |
| anti-β-Actin-HRP | WB | 0.1 µg/mL | sc-47778 HRP, Santa Cruz Biotechnology (Dallas, TX, USA) |
| anti-Rabbit-Alexa-488 | IS | 5 µg/mL | A27034, ThermoFisher (Waltham, MA, USA) |
| Anti-LAMP2-Alexa-488 | IS | 5 µg/mL | 108510, BioLegend (San Diego, CA, USA) |
| DAPI | IS | 20 µg/mL | D1306, Thermo Fisher (Waltham, MA, USA) |
| Texas Red-X Phalloidin | IS | 5 µg/mL | T7472, Thermo Fisher (Waltham, MA, USA) |
| Identified Protein | Protein ID | No of Unique Peptide | Identified/Total Aminoacids |
|---|---|---|---|
| Adenosine receptor 2A | Q60613 | 6 | 52/410 |
| Coatomer subunit gamma-2 | Q9QXK3 | 1 | 29/871 |
| Niemann–Pick C1 protein | O35604 | 2 | 24/1278 |
| Isoform 2 of splicing factor 3B subunit 3 | Q921M3–2 | 2 | 27/1122 |
| Sec1 family domain-containing protein 1 | Q8BRF7 | 2 | 34/639 |
| Chaperone protein DnaJ | Q3TK61 | 2 | 48/397 |
| Thyroid hormone receptor-associated protein | Q569Z6 | 1 | 19/951 |
| P2X7 purinoceptor | J7IR93 | 3 | 34/366 |
| Cathepsin D | P18242 | 2 | 36/410 |
| Monoacylglycerol lipase ABHD12n | Q99LR1 | 2 | 23/398 |
| AP-3 complex subunit mu-1 | H7BWY2 | 1 | 26/364 |
| Ras-related protein Rab 18 | P35293 | 2 | 28/206 |
| RUN and FYVE domain containing protein | Q8BIJ7 | 1 | 14/712 |
| Myeloid cell nuclear differentiation antigen-like protein | D0QMC3 | 1 | 12/538 |
| Caprin-1 | Q60865 | 1 | 40/707 |
| Myof protein | B9EK95 | 3 | 37/2061 |
| DNA topoisomerase 1 | Q04750 | 3 | 35/767 |
| Alpha glucosidase 2 alpha neutral subunit | A1A4T2 | 2 | 27/966 |
| Receptor mediated endocytosis-8 | D4AFX7 | 1 | 15/2248 |
| T-complex protein 1 subunit gamma | E9Q133 | 2 | 24/507 |
| Heterogeneous nuclear ribonucleoprotein U-like protein 2 | Q00PI9 | 2 | 23/745 |
| Elongation factor Tu | D3YVN7 | 2 | 35/452 |
| DEAD (Asp-Glu-Ala-Asp) box polypeptide 21 | Q6PCP0 | 2 | 37/851 |
| Annexin A5 | P48046 | 2 | 31/319 |
| O-acyltransferase | Q06EZ3 | 1 | 9/540 |
| Serine/threonine-protein phosphatase | Q8BN07 | 1 | 11/285 |
| Histone deacetylase | D3YYI8 | 1 | 12/482 |
| Coronin-1B | Q9WUM3 | 2 | 24/484 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skopál, A.; Ujlaki, G.; Gerencsér, A.T.; Bankó, C.; Bacsó, Z.; Ciruela, F.; Virág, L.; Haskó, G.; Kókai, E. Adenosine A2A Receptor Activation Regulates Niemann–Pick C1 Expression and Localization in Macrophages. Curr. Issues Mol. Biol. 2023, 45, 4948-4969. https://doi.org/10.3390/cimb45060315
Skopál A, Ujlaki G, Gerencsér AT, Bankó C, Bacsó Z, Ciruela F, Virág L, Haskó G, Kókai E. Adenosine A2A Receptor Activation Regulates Niemann–Pick C1 Expression and Localization in Macrophages. Current Issues in Molecular Biology. 2023; 45(6):4948-4969. https://doi.org/10.3390/cimb45060315
Chicago/Turabian StyleSkopál, Adrienn, Gyula Ujlaki, Attila Tibor Gerencsér, Csaba Bankó, Zsolt Bacsó, Francisco Ciruela, László Virág, György Haskó, and Endre Kókai. 2023. "Adenosine A2A Receptor Activation Regulates Niemann–Pick C1 Expression and Localization in Macrophages" Current Issues in Molecular Biology 45, no. 6: 4948-4969. https://doi.org/10.3390/cimb45060315
APA StyleSkopál, A., Ujlaki, G., Gerencsér, A. T., Bankó, C., Bacsó, Z., Ciruela, F., Virág, L., Haskó, G., & Kókai, E. (2023). Adenosine A2A Receptor Activation Regulates Niemann–Pick C1 Expression and Localization in Macrophages. Current Issues in Molecular Biology, 45(6), 4948-4969. https://doi.org/10.3390/cimb45060315