
How Do Cancer-Related Mutations Affect the Oligomerisation State of the p53 Tetramerisation Domain?
 , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of the 37TDs
2.1.1. Peptide Purification
2.1.2. Characterisation
2.2. Circular Dichroism
2.3. Native Mass Spectrometry
2.4. Nuclear Magnetic Resonance (NMR)
3. Results and Discussion
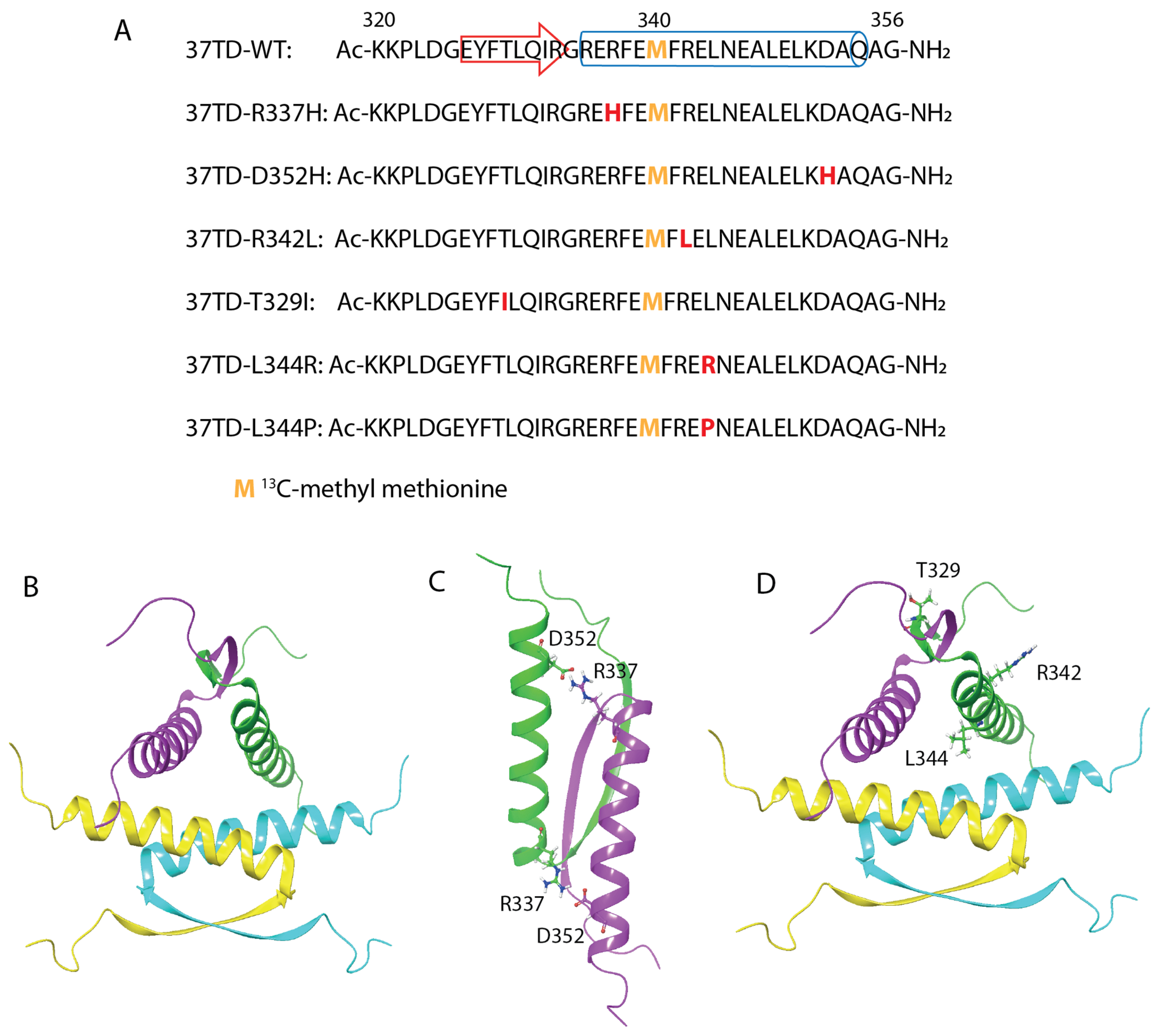
3.1. Definition of the TD and Selection of the Mutations

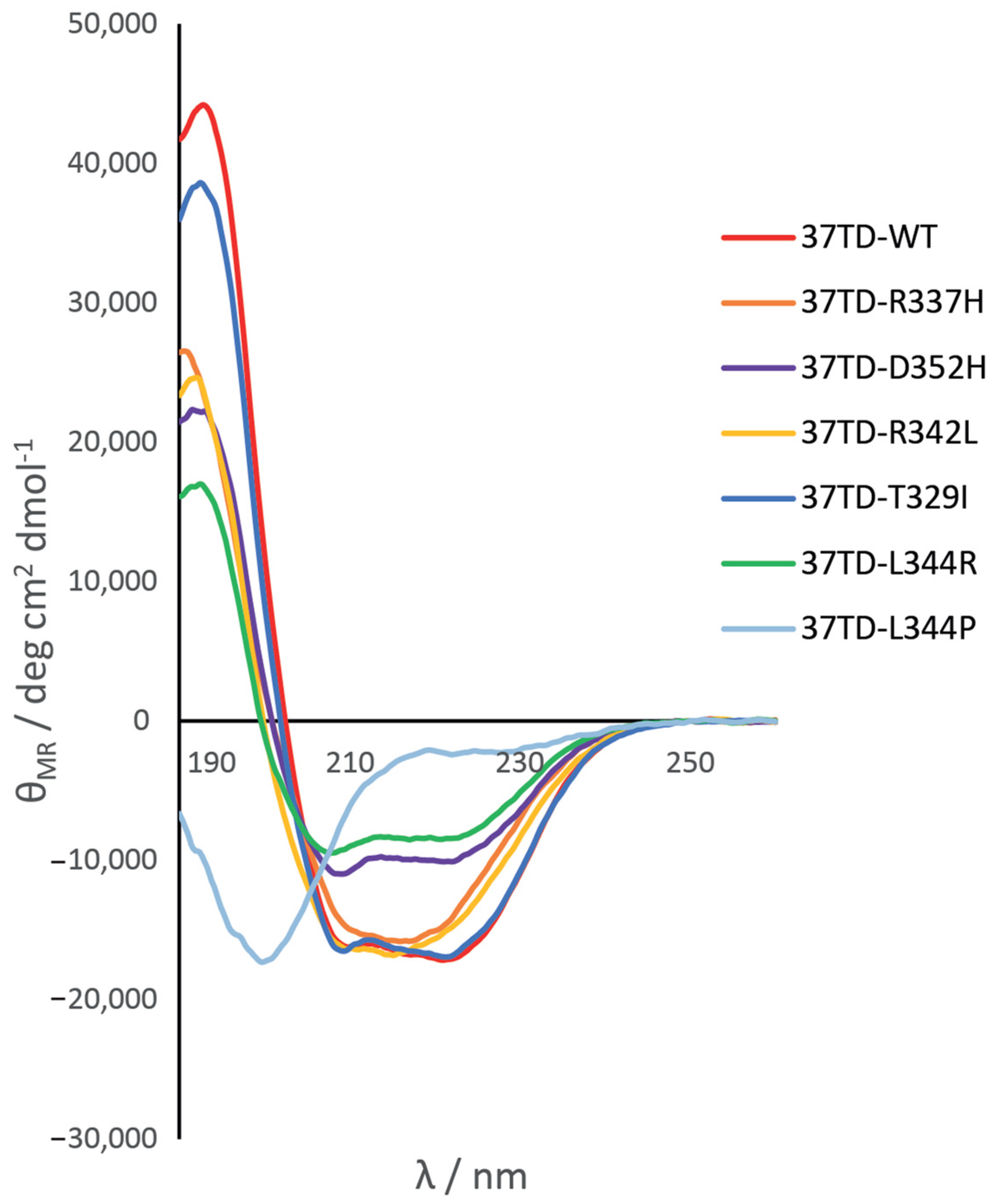
3.2. Secondary Structure of the 37TDs
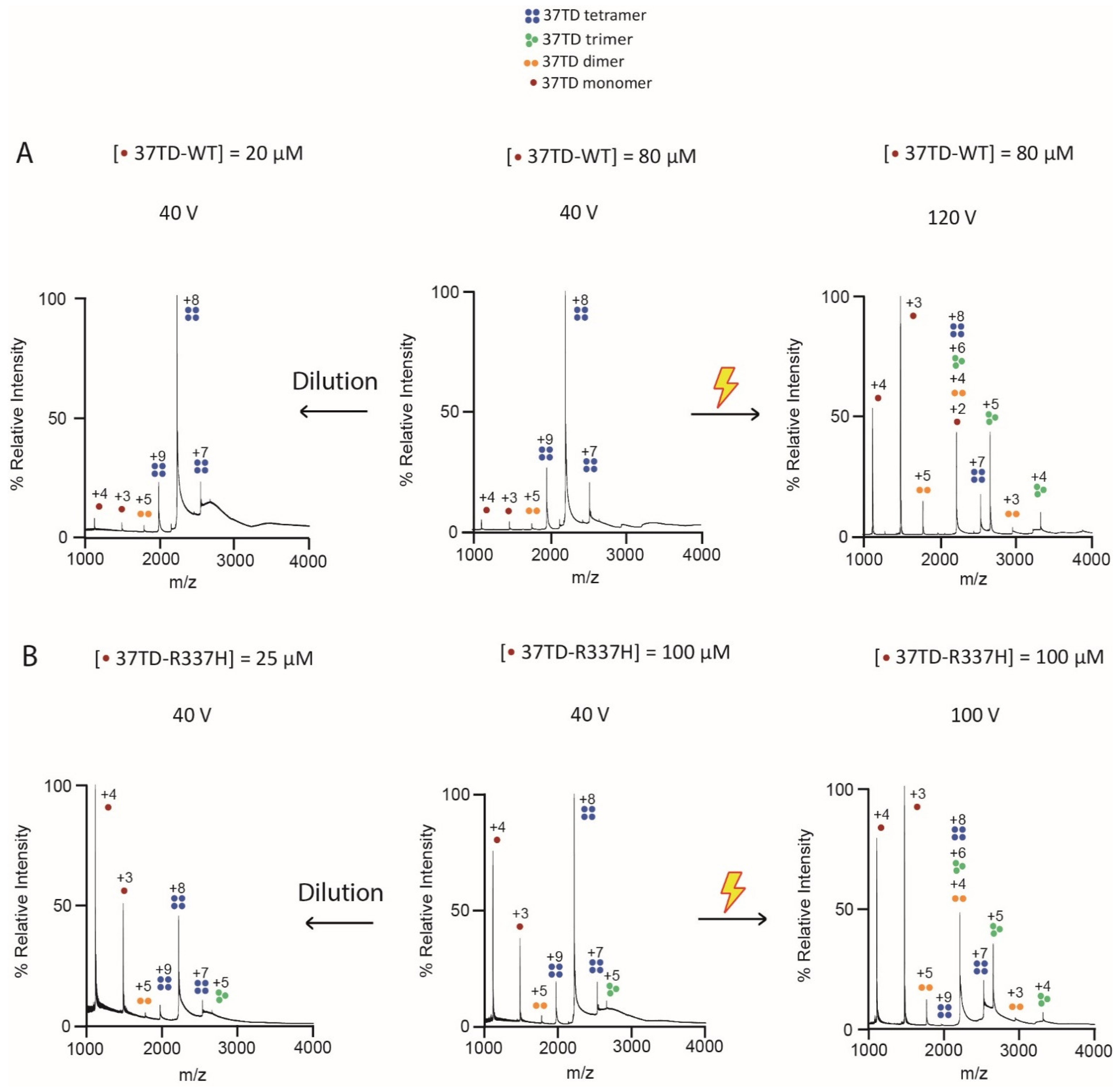
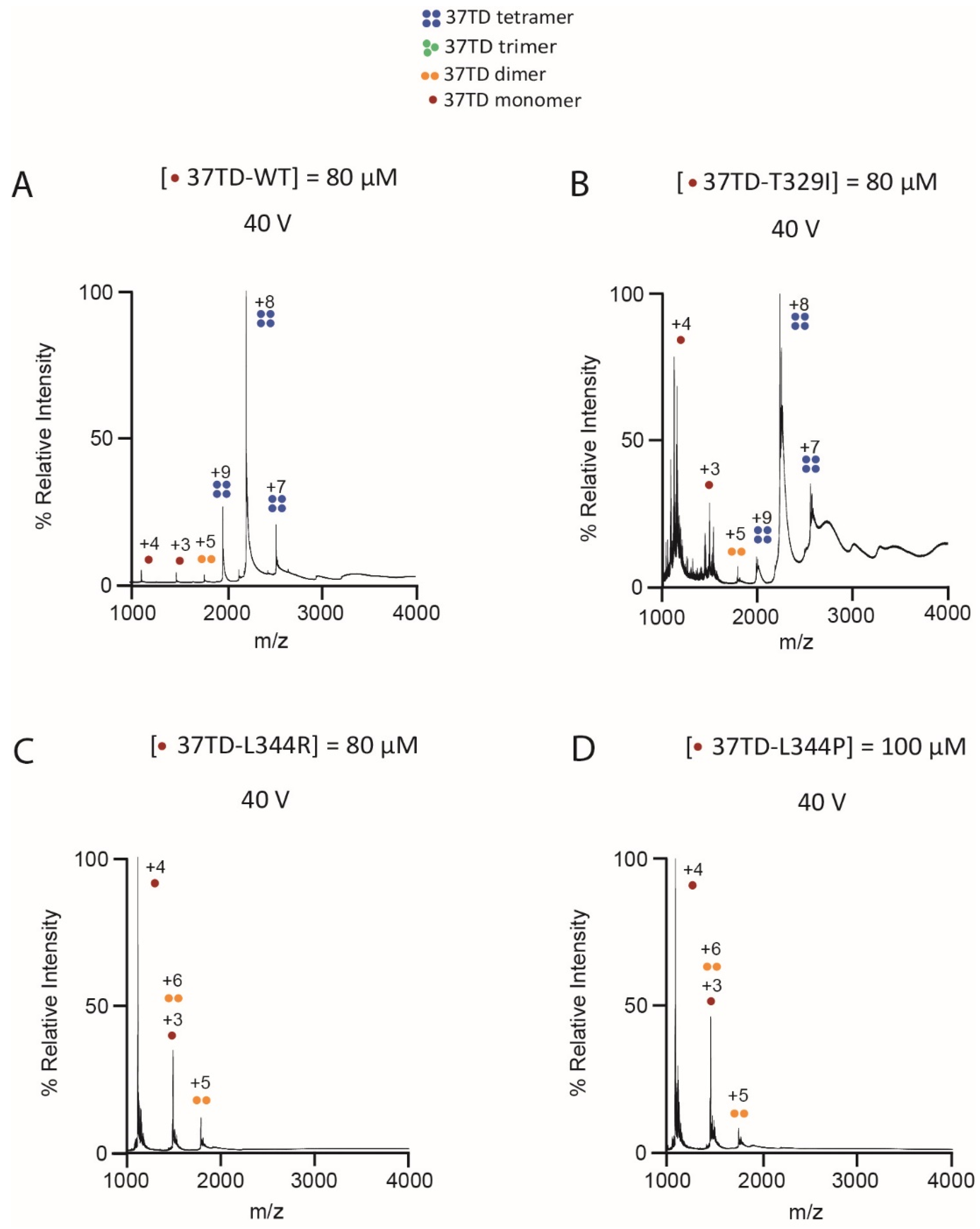
3.3. Determination of the Oligomeric State by Native MS
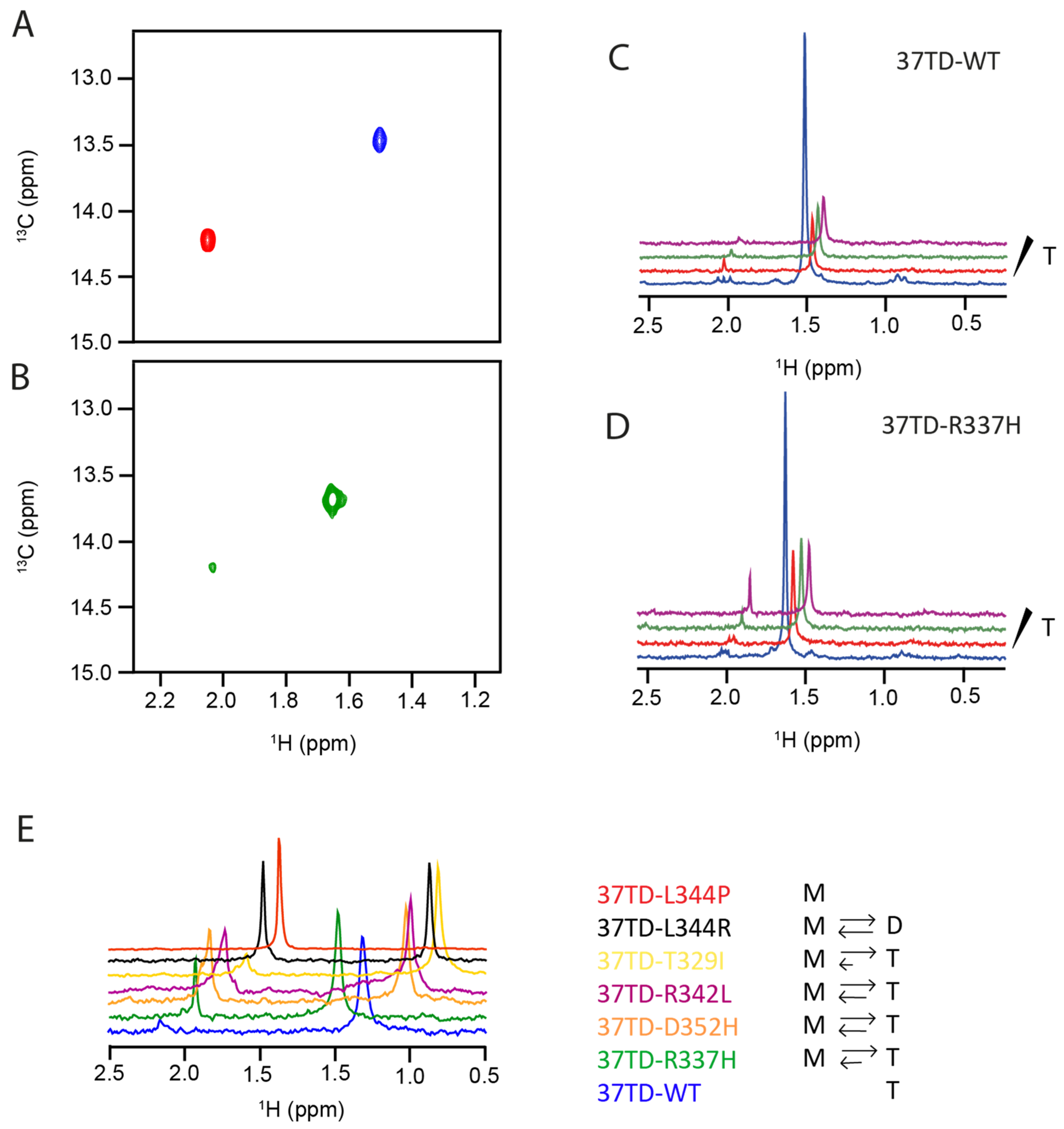
3.4. Study of the Equilibrium between Oligomers by NMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffens, L.; Groen, K.; Newton, C.; Avery-kiejda, K.A. The role of truncated p53 isoforms in the DNA damage response. Biochim. Biophys. Acta BBA Rev. Cancer 2023, 1878, 188882. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Huang, F.; Fersht, A.R. Single-molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2010, 39, 2294–2303. [Google Scholar] [CrossRef] [Green Version]
- Gencel-Augusto, J.; Su, X.; Qi, Y.; Whitley, E.M.; Pant, V.; Xiong, S.; Shah, V.; Lin, J.; Perez, E.; Fiorotto, M.L.; et al. Dimeric p53 Mutant Elicits Unique Tumor-Suppressive Activities through an Altered Metabolic Program. Cancer Discov. 2023, 13, 1230–1249. [Google Scholar] [CrossRef]
- Gaglia, G.; Guan, Y.; Shah, J.V.; Lahav, G. Activation and control of p53 tetramerization in individual living cells. Proc. Natl. Acad. Sci. USA 2013, 110, 15497–15501. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, L.M.M.; Durell, S.R.; Mazur, S.J.; Appella, E. p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Wang, D. Deciphering the PTM codes of the tumor suppressor p53. J. Mol. Cell Biol. 2021, 13, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; Hall, P.A. The P53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Okorokov, A.L.; Sherman, M.B.; Plisson-Chastang, C.; Grinkevich, V.; Sigmundsson, K.; Selivanova, G.; Milner, J.; Orlova, E.V. The structure of p53 tumour suppressor protein reveals the basis for its functional plasticity. EMBO J. 2006, 25, 5191–5200. [Google Scholar] [CrossRef] [Green Version]
- Bista, M.; Freund, S.M.; Fersht, A.R. Domain–domain interactions in full-length p53 and a specific DNA complex probed by methyl NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 15752–15756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, N.W.; Prodeus, A.; Tran, J.; Malkin, D.; Gariépy, J. Association between the oligomeric status of p53 and clinical outcomes in Li-Fraumeni syndrome. Gynecol. Oncol. 2018, 110, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Rigoli, M.; Spagnolli, G.; Lorengo, G.; Monti, P.; Potestio, R.; Biasini, E.; Inga, A. Structural Basis of Mutation-Dependent p53 Tetramerization Deficiency. Int. J. Mol. Sci. 2022, 23, 7960. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Wang, J.; Liu, J. p53 regulation by ubiquitin and ubiquitin-like modifications. Genome Instab. Dis. 2022, 3, 179–198. [Google Scholar] [CrossRef]
- D’Abramo, M.; Besker, N.; Desideri, A.; Levine, A.J.; Melino, G.; Chillemi, G. The p53 tetramer shows an induced-fit interaction of the C-terminal domain with the DNA-binding domain. Oncogene 2015, 35, 3272–3281. [Google Scholar] [CrossRef]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhou, X.; Wang, J.; Li, Z.; Kong, X.; Qian, J.; Hu, Y.; Fang, J.-Y. RhoGAPs Attenuate Cell Proliferation by Direct Interaction with p53 Tetramerization Domain. Cell Rep. 2013, 3, 1526–1538. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Yue, J.; Liu, Z.; Shen, Z. Abrogation of the transactivation activity of p53 by BCCIP down-regulation. J. Biol. Chem. 2007, 282, 1570–1576. [Google Scholar] [CrossRef] [Green Version]
- Kuusk, A.; Boyd, H.; Chen, H.; Ottmann, C. Small-molecule modulation of p53 protein-protein interactions. Biol. Chem. 2020, 401, 921–931. [Google Scholar] [CrossRef]
- Lee, I.H.; Kawai, Y.; Fergusson, M.M.; Rovira, I.I.; Bishop, A.J.R.; Motoyama, N.; Cao, L.; Finkel, T. Atg7 Modulates p53 Activity to Regulate Cell Cycle and Survival during Metabolic Stress. Science 2012, 336, 225–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, W.; Hayashi, Y.; Yokoyama, W.; Kuroda, T.; Kishimoto, H.; Ito, I.; Kimura, K.; Akaogi, K.; Waku, T.; Yanagisawa, J. The nucleolar protein Myb-binding protein 1A (MYBBP1A) enhances p53 tetramerization and acetylation in response to nucleolar disruption. J. Biol. Chem. 2014, 289, 4928–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, K.; Sheikh, M.S.; Huang, Y. Regulation of p53 oligomerization by Ras superfamily protein RBEL1A. Genes Cancer 2015, 6, 307–316. Available online: http://www.impactjournals.com/ (accessed on 20 July 2021). [CrossRef] [PubMed] [Green Version]
- van Dieck, J.; Fernandez-Fernandez, M.R.; Veprintsev, D.; Fersht, A.R. Modulation of the Oligomerization State of p53 by Differential Binding of Proteins of the S100 Family to p53 Monomers and Tetramers. J. Biol. Chem. 2009, 284, 13804–13811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foo, R.S.-Y.; Nam, Y.-J.; Ostreicher, M.J.; Metzl, M.D.; Whelan, R.S.; Peng, C.-F.; Ashton, A.W.; Fu, W.; Mani, K.; Chin, S.-F.; et al. Regulation of p53 tetramerization and nuclear export by ARC. Proc. Natl. Acad. Sci. USA 2007, 104, 20826–20831. [Google Scholar] [CrossRef] [Green Version]
- Katz, C.; Low-Calle, A.M.; Choe, J.H.; Laptenko, O.; Tong, D.; Joseph-Chowdhury, J.-S.N.; Garofalo, F.; Zhu, Y.; Friedler, A.; Prives, C. Wild-type and cancer-related p53 proteins are preferentially degraded by MDM2 as dimers rather than tetramers. Genes Dev. 2018, 32, 430–447. [Google Scholar] [CrossRef] [Green Version]
- Ano Bom, A.P.D.; Rangel, L.P.; Costa, D.C.F.; de Oliveira, G.A.P.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.; Cepeda, A.O.T.; Stumbo, A.; et al. Mutant p53 Aggregates into Prion-like Amyloid Oligomers and Fibrils: Implications for cancer. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, R.; Nomura, T.; Anderson, C.W.; Sakaguchi, K. Cancer-associated p53 tetramerization domain mutants: Quantitative analysis reveals a low threshold for tumor suppressor inactivation. J. Biol. Chem. 2011, 286, 252–258. [Google Scholar] [CrossRef] [Green Version]
- Senitzki, A.; Safieh, J.; Sharma, V.; Golovenko, D.; Danin-Poleg, Y.; Inga, A.; E Haran, T. The complex architecture of p53 binding sites. Nucleic Acids Res. 2021, 49, 1364–1382. [Google Scholar] [CrossRef]
- Gencel-Augusto, J.; Lozano, G. p53 tetramerization: At the center of the dominant-negative effect of mutant p53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, T.; Kato, S.; Otsuka, K.; Watanabe, G.; Kumabe, T.; Tominaga, T.; Yoshimoto, T.; Ishioka, C. The relationship among p53 oligomer formation, structure and transcriptional activity using a comprehensive missense mutation library. Oncogene 2005, 24, 6976–6981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imagawa, T.; Terai, T.; Yamada, Y.; Kamada, R.; Sakaguchi, K. Evaluation of transcriptional activity of p53 in individual living mammalian cells. Anal. Biochem. 2009, 387, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Li, J.; Starost, M.F.; Liu, C.; Zhuang, J.; Chen, J.; Achatz, M.I.; Kang, J.-G.; Wang, P.-Y.; Savage, S.A.; et al. Mouse Homolog of the Human TP53 R337H Mutation Reveals Its Role in Tumorigenesis. Cancer Res. 2018, 78, 5375–5383. [Google Scholar] [CrossRef] [Green Version]
- Fischer, N.W.; Prodeus, A.; Malkin, D.; Gariépy, J. p53 oligomerization status modulates cell fate decisions between growth, arrest and apoptosis. Cell Cycle 2016, 15, 3210–3219. [Google Scholar] [CrossRef] [Green Version]
- Giacomelli, A.O.; Yang, X.; Lintner, R.E.; McFarland, J.M.; Duby, M.; Kim, J.; Howard, T.P.; Takeda, D.Y.; Ly, S.H.; Kim, E.; et al. Mutational processes shape the landscape of TP53 mutations in human cancer HHS Public Access Author manuscript. Nat. Genet. 2018, 50, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Mateu, M.G. Nine hydrophobic side chains are key determinants of the thermodynamic stability and oligomerization status of tumour suppressor p53 tetramerization domain. EMBO J. 1998, 17, 2748–2758. [Google Scholar] [CrossRef] [Green Version]
- Eissler, S.; Kley, M.; Bächle, D.; Loidl, G.; Meier, T.; Samson, D. Substitution determination of Fmoc-substituted resins at different wavelengths. J. Pept. Sci. 2017, 23, 757–762. [Google Scholar] [CrossRef]
- Kreutzer, A.G.; Salveson, P.J. Standard Practices for Fmoc-Based Solid-Phase Peptide Synthesis in the Nowick Laboratory (Version 1.6.1). p. 2015. Available online: https://docplayer.net/9616568-Standard-practices-for-fmoc-based-solid-phase-peptide-synthesis-in-the-nowick-laboratory-version-1-6-1.html (accessed on 20 July 2021).
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Christensen, T. A Qualitative Test for Monitoring Coupling Completeness in Solid Phase Peptide Synthesis Using Chloranil. Acta Chem. Scand. 1979, 33, 763–766. [Google Scholar] [CrossRef]
- Palasek, S.A.; Cox, Z.J.; Collins, J.M. Limiting racemization and aspartimide formation in microwave-enhanced Fmoc solid phase peptide synthesis. J. Pept. Sci. 2007, 13, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Hopper, J.T.S.; Oldham, N.J. Collision Induced Unfolding of Protein Ions in the Gas Phase Studied by Ion Mobility-Mass Spectrometry: The Effect of Ligand Binding on Conformational Stability. J. Am. Soc. Mass Spectrom. 2009, 20, 1851–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.P.; Knapman, T.W.; Campuzano, I.; Malham, R.W.; Berryman, J.T.; Radford, S.E.; Ashcroft, A.E. Deciphering drift time measurements from travelling wave ion mobility spectrometry-Mass spectrometry studies. Eur. J. Mass Spectrom. 2009, 15, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Ruotolo, B.T.; Benesch, J.L.P.; Sandercock, A.M.; Hyung, S.-J.; Robinson, C.V. Ion mobility–mass spectrometry analysis of large protein complexes. Nat. Protoc. 2008, 3, 1139–1152. [Google Scholar] [CrossRef] [Green Version]
- Rubinson, K.A. Practical corrections for p(H,D) measurements in mixed H2O/D2O biological buffers. Anal. Methods 2017, 9, 2744–2750. [Google Scholar] [CrossRef]
- Ferrage, F.; Zoonens, M.; Warschawski, D.E.; Popot, J.L.; Bodenhausen, G. Slow diffusion of macromolecular assemblies by a new pulsed field gradient NMR method. J. Am. Chem. Soc. 2003, 125, 2541–2545. [Google Scholar] [CrossRef]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef]
- Marsh, J.A.; Forman-Kay, J.D. Sequence Determinants of Compaction in Intrinsically Disordered Proteins. Biophys. J. 2010, 98, 2383–2390. [Google Scholar] [CrossRef] [Green Version]
- Tidow, H.; Melero, R.; Mylonas, E.; Freund, S.M.V.; Grossmann, J.G.; Carazo, J.M.; Svergun, D.I.; Valle, M.; Fersht, A.R. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc. Natl. Acad. Sci. USA 2007, 104, 12324–12329. [Google Scholar] [CrossRef] [Green Version]
- Pindado, J.G. Synthesis of Biaryl Bicyclic Peptides for Recognition of Protein Surfaces. Universitat de Barcelona. May 2017. Available online: http://www.tesisenred.net/handle/10803/403399 (accessed on 5 November 2021).
- Villoslada, S.G. Use of Calix[4]arenes to Recover the Self-Assembly Ability of Mutated p53 Tetramerization Domains. Universitat de Barcelona. 2008. Available online: http://www.tesisenred.net/handle/10803/2819 (accessed on 5 November 2021).
- Clore, G.M.; Omichinski, J.G.; Sakaguchi, K.; Zambrano, N.; Sakamoto, H.; Appella, E.; Gronenborn, A.M. High-Resolution Structure of the Oligomerization Domain of p53 by Multidimensional NMR. Science 1994, 265, 386–391. [Google Scholar] [CrossRef]
- DiGiammarino, E.L.; Lee, A.S.; Cadwell, C.; Zhang, W.; Bothner, B.; Ribeiro, R.C.; Zambetti, G.; Kriwacki, R.W. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat. Struct. Biol. 2001, 9, 12–16. [Google Scholar] [CrossRef]
- Gordo, S.; Martos, V.; Santos, E.; Menéndez, M.; Bo, C.; Giralt, E.; de Mendoza, J. Stability and structural recovery of the tetramerization domain of p53-R337H mutant induced by a designed templating ligand. Proc. Natl. Acad. Sci. USA 2008, 105, 16426–16431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, E.M.; Billerbeck, A.E.C.; Villares, M.C.B.F.; Domenice, S.; Mendonça, B.B.; Latronico, A.C.A. Founder Effect for the Highly Prevalent R337H Mutation ofTumor Suppressor p53 in Brazilian Patients with Adrenocortical Tumors. Arq. Bras Endocrinol. Metab. 2004, 48, 647–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Dong, X.; Tang, Y.; Li, L.; Wei, G. Mechanistic insight into the destabilization of p53TD tetramer by cancer-related R337H mutation: A molecular dynamic study. Phys. Chem. Chem. Phys. 2022, 24, 5199–5210. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.M.; Zambetti, G. What 20 Years of Research has Taught us about the TP53 p.R337H Mutation. Cancer 2020, 126, 4678–4686. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, J.R.; Pinto, E.M.; Rehg, J.E.; Clay, M.R.; Wang, J.; Neale, G.; Heath, R.J.; Lozano, G.; Lalli, E.; Figueiredo, B.C.; et al. The common germline TP53-R337H mutation is hypomorphic and confers incomplete penetrance and late tumor onset in a mouse model. Cancer Res 2021, 81, 2442–2456. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Leney, A.C.; Heck, A.J.R. Native Mass Spectrometry: What is in the Name? J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. [Google Scholar] [CrossRef] [Green Version]
- May, J.C.; McLean, J.A. Ion mobility-mass spectrometry: Time-dispersive instrumentation. Anal. Chem. 2015, 87, 1422–1436. [Google Scholar] [CrossRef] [Green Version]
- Jurchen, J.C.; Williams, E.R. Origin of asymmetric charge partitioning in the dissociation of gas-phase protein homodimers. J. Am. Chem. Soc. 2003, 125, 2817–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciudad, S.; Puig, E.; Botzanowski, T.; Meigooni, M.; Arango, A.S.; Do, J.; Mayzel, M.; Bayoumi, M.; Chaignepain, S.; Maglia, G.; et al. Aβ(1-42) tetramer and octamer structures reveal edge conductivity pores as a mechanism for membrane damage. Nat. Commun. 2020, 11, 3014. [Google Scholar] [CrossRef] [PubMed]
- López, A.; Tarragó, T.; Vilaseca, M.; Giralt, E. Applications and future of ion mobility mass spectrometry in structural biology. New J. Chem. 2013, 37, 1283–1289. [Google Scholar] [CrossRef]
- Mora, P.; Carbajo, R.J.; Pineda-Lucena, A.; del Pino, M.M.S.; Pérez-Payá, E. Solvent-exposed residues located in the β-sheet modulate the stability of the tetramerization domain of p53-A structural and combinatorial approach. Proteins: Struct. Funct. Genet. 2008, 71, 1670–1685. [Google Scholar] [CrossRef]
- Wishart, D.S.; Bigam, C.G.; Holm, A.; Hodges, R.S.; Sykes, B.D. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 1995, 5, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source T | Bias | Trap | Transfer | Backing | Wave Velocity (IM) | Wave Height (IM) |
|---|---|---|---|---|---|---|
| 40 °C | 4 V (TOF) | 6 V | 4 V | 5.7–5.9 mbar | 300 m/s | 8 V |
| 15 V (IM) |
| Residues | Reference |
|---|---|
| 325–353 | Bista et al. [12] |
| 325–355 | Rajagopalan et al. [5] |
| 326–356 | Gaglia et al. [7] |
| 323–360 | Tidow et al. [51] |
| 320–356 | García [52], Gordo [53] |
| 319–358 | Kamada et al. [29] |
| 37TDs | D × 10−10/m2 s−1 | RH/Å Experimental | RH/Å Predicted |
|---|---|---|---|
| 37TD-WT | 1.00 ± 0.02 | 22.2 | 20.3 a |
| 37TD-L344P | 1.45 ± 0.01 | 15.3 | 14.6 b |
| 37TD-L344R Dimer Monomer | |||
| 1.38 ± 0.02 | 16.1 | 16.5 c | |
| 1.78 ± 0.04 | 12.5 | 14.1 b | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolini, F.; Todorovski, T.; Puig, E.; Díaz-Lobo, M.; Vilaseca, M.; García, J.; Andreu, D.; Giralt, E. How Do Cancer-Related Mutations Affect the Oligomerisation State of the p53 Tetramerisation Domain? Curr. Issues Mol. Biol. 2023, 45, 4985-5004. https://doi.org/10.3390/cimb45060317
Nicolini F, Todorovski T, Puig E, Díaz-Lobo M, Vilaseca M, García J, Andreu D, Giralt E. How Do Cancer-Related Mutations Affect the Oligomerisation State of the p53 Tetramerisation Domain? Current Issues in Molecular Biology. 2023; 45(6):4985-5004. https://doi.org/10.3390/cimb45060317
Chicago/Turabian StyleNicolini, Federica, Toni Todorovski, Eduard Puig, Mireia Díaz-Lobo, Marta Vilaseca, Jesús García, David Andreu, and Ernest Giralt. 2023. "How Do Cancer-Related Mutations Affect the Oligomerisation State of the p53 Tetramerisation Domain?" Current Issues in Molecular Biology 45, no. 6: 4985-5004. https://doi.org/10.3390/cimb45060317
APA StyleNicolini, F., Todorovski, T., Puig, E., Díaz-Lobo, M., Vilaseca, M., García, J., Andreu, D., & Giralt, E. (2023). How Do Cancer-Related Mutations Affect the Oligomerisation State of the p53 Tetramerisation Domain? Current Issues in Molecular Biology, 45(6), 4985-5004. https://doi.org/10.3390/cimb45060317