
Dimethyloxalylglycine Suppresses SREBP1c and Lipogenic Gene Expressions in Hepatocytes Independently of HIF1A



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Lysis, Subcellular Fractionation, and Immunoblot Analysis
2.3. Total RNA Isolation and Reverse Transcription-Quantitative PCR
2.4. Transfection and Luciferase Assays
2.5. RNA Interference
2.6. Animal Studies
2.7. Statistical Analysis
3. Results
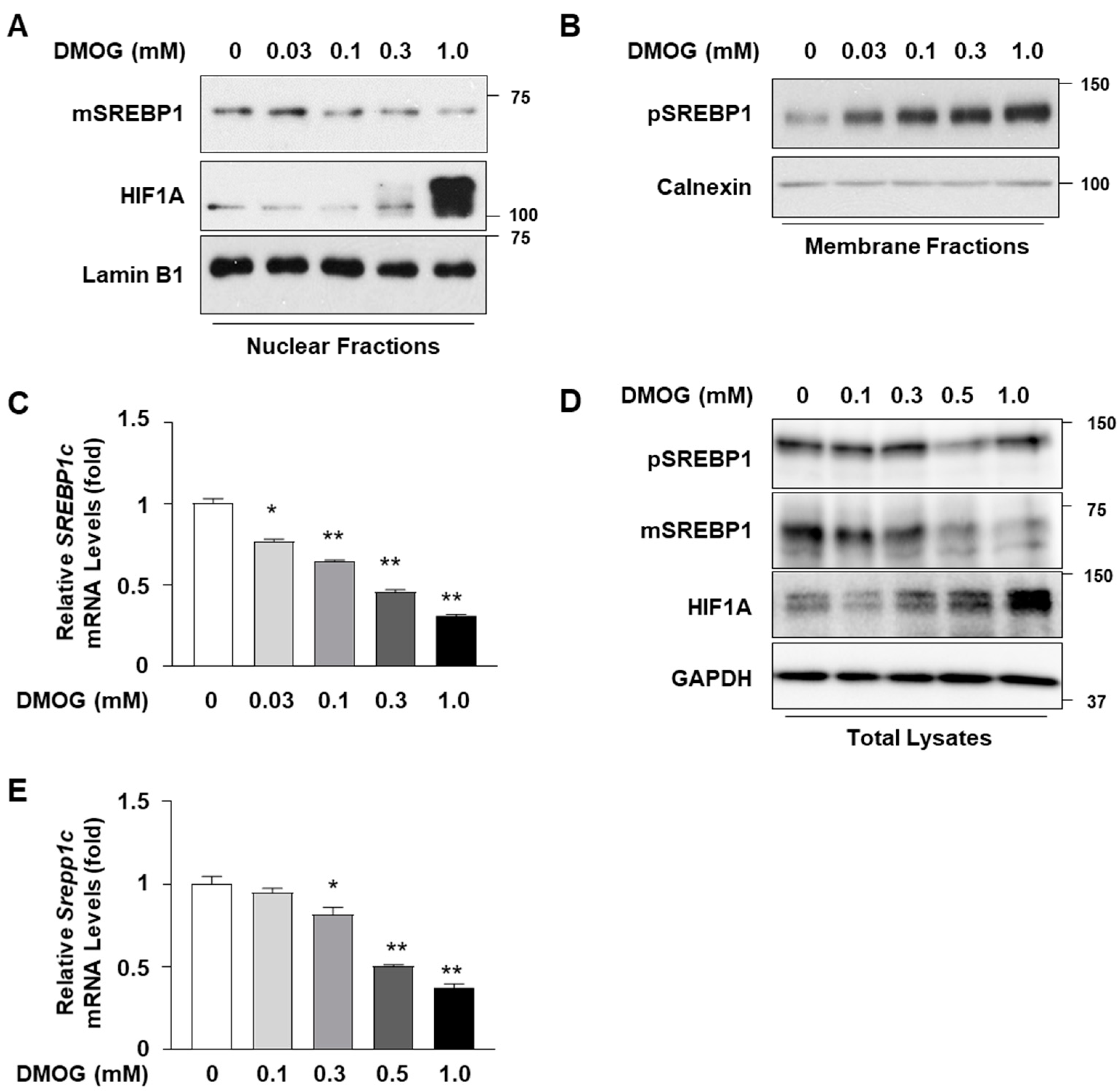
3.1. DMOG Represses SREBP1c Expression in Hepatocytes
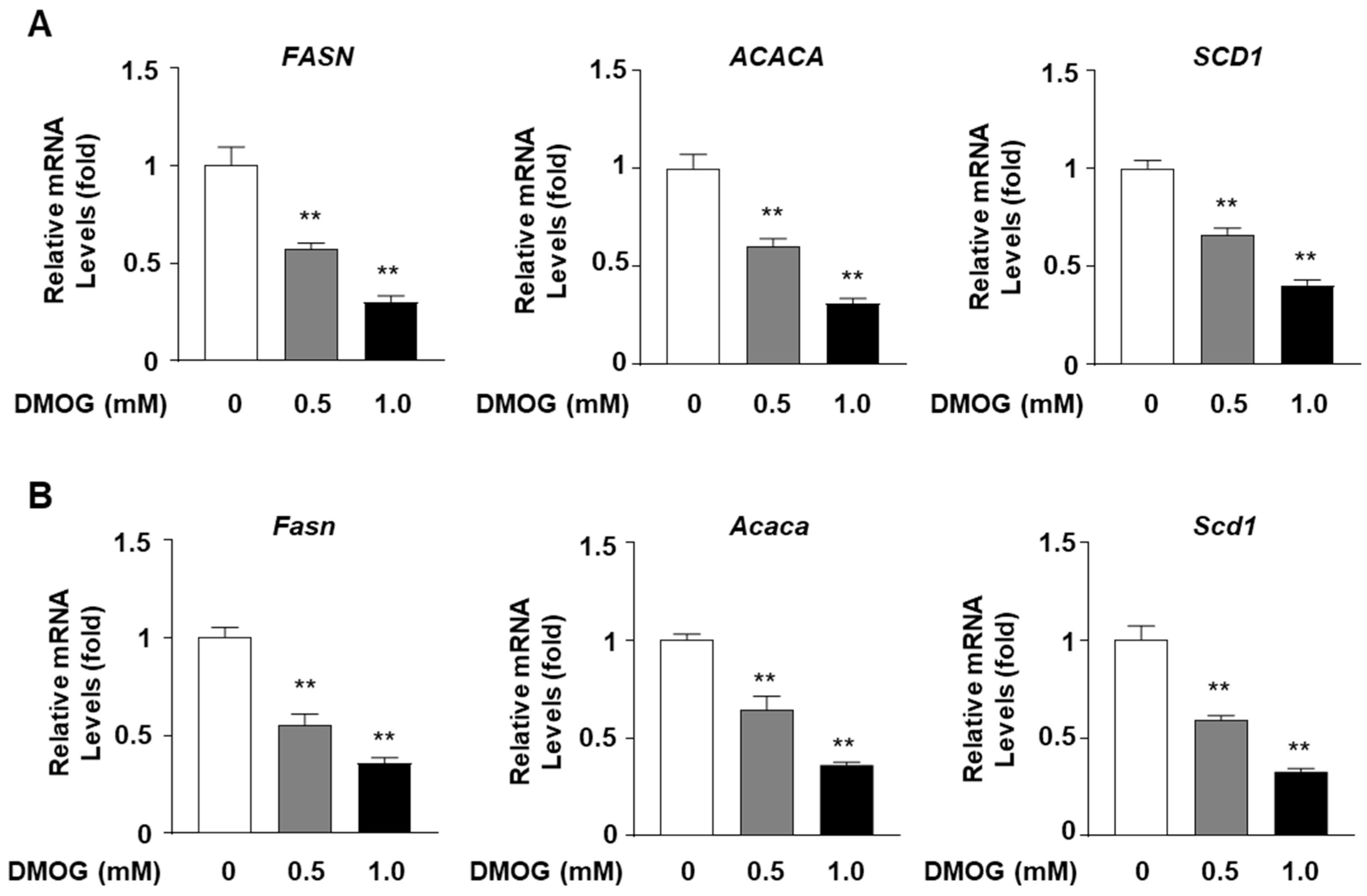
3.2. DMOG Inhibits the Expression of SREBP1-Regulated Lipogenic Genes
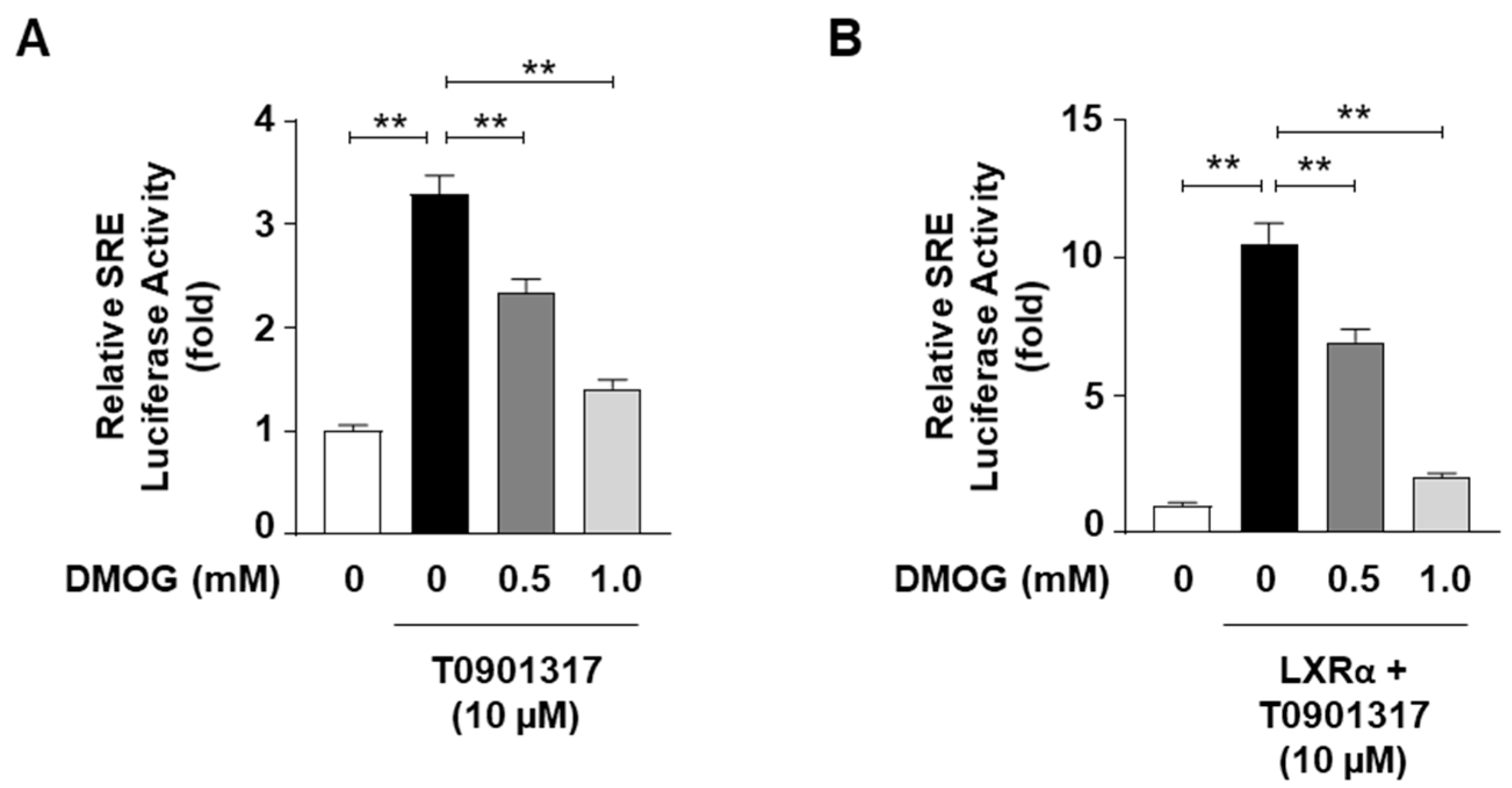
3.3. DMOG Inhibits the Transcriptional Activity of SREBP1c
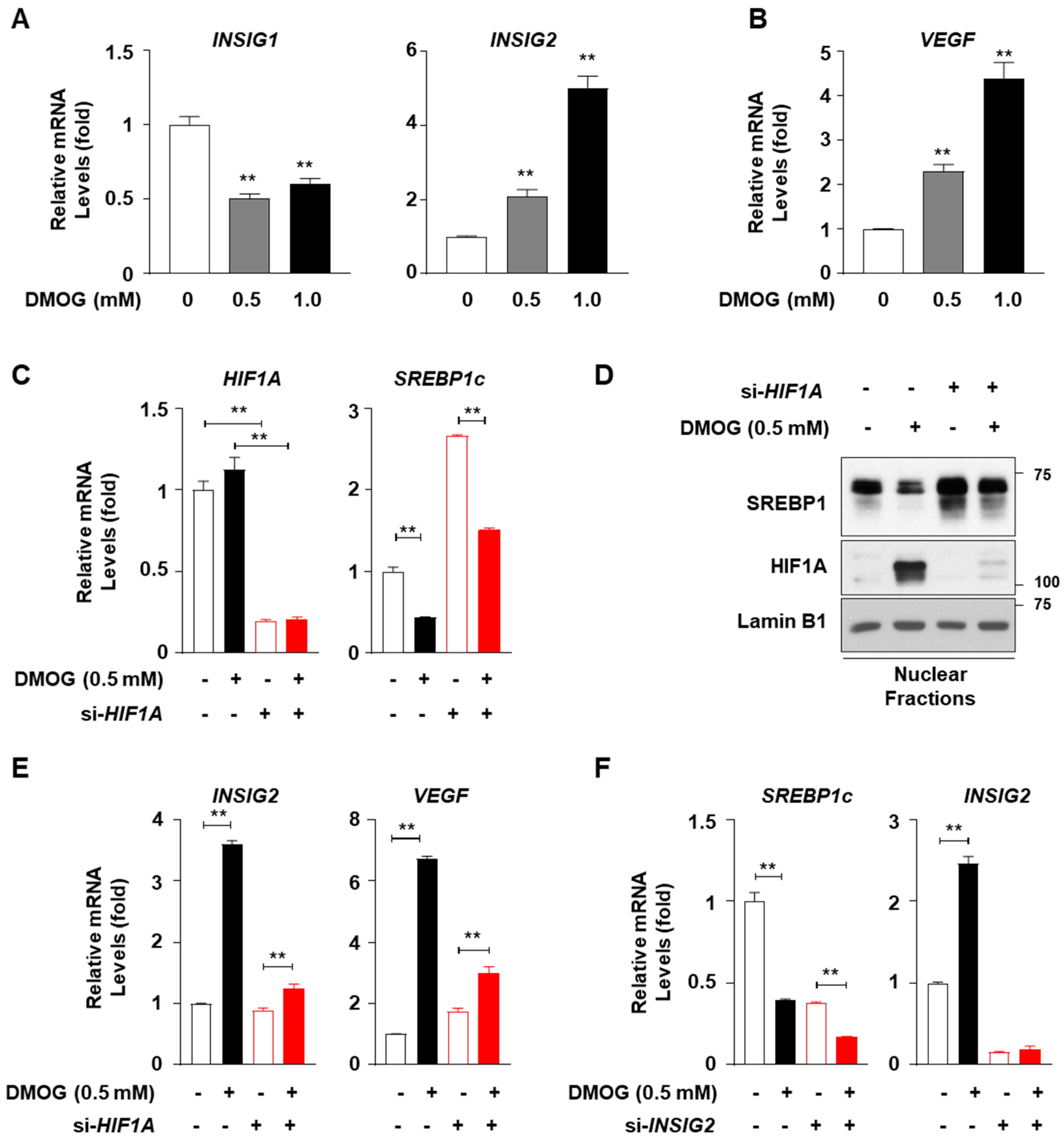
3.4. Inhibitory Effect of DMOG on SREBP1c Expression Is Independent of HIF1A and INSIG2
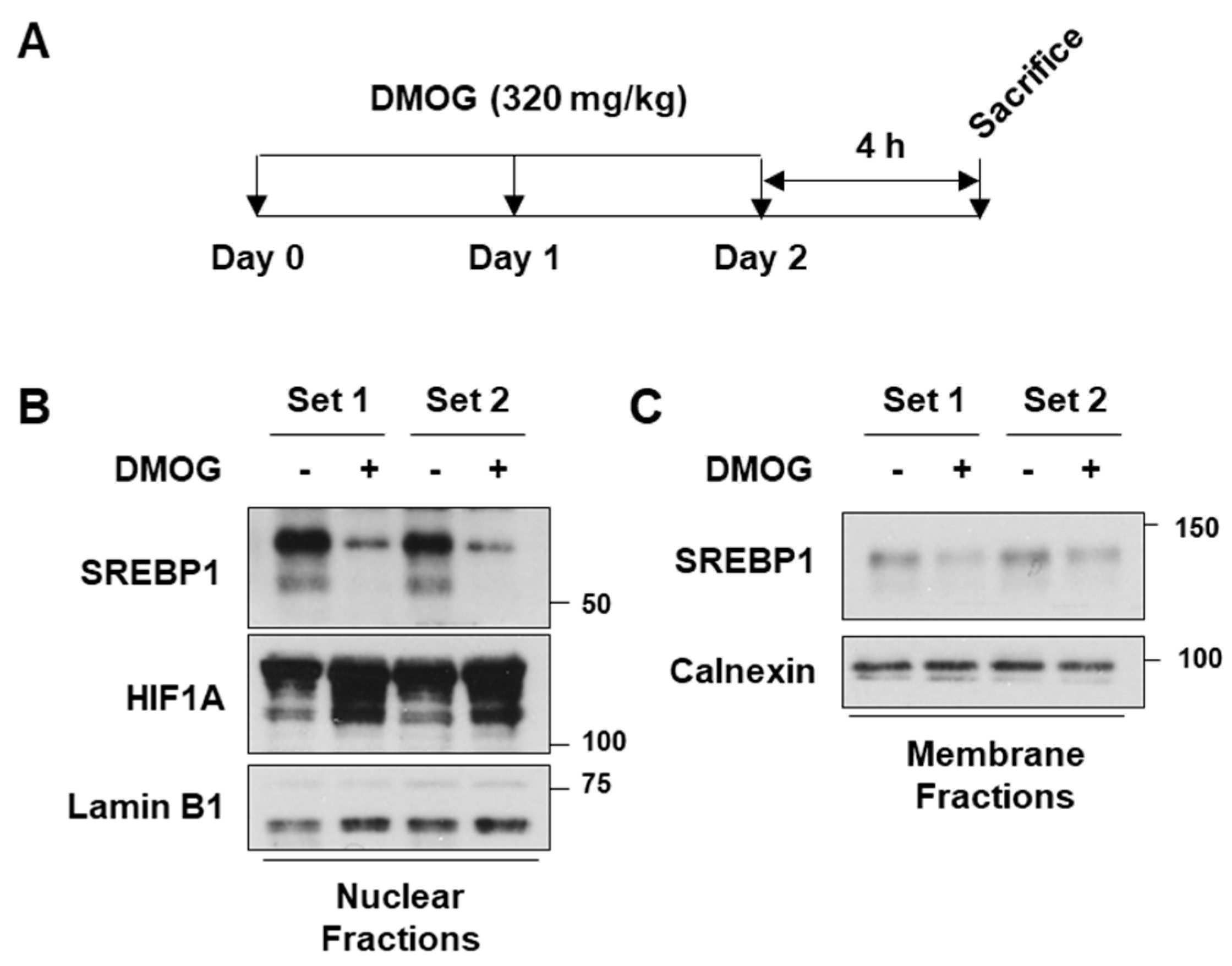
3.5. DMOG Reduces SREBP1c Expression in Mice Liver
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ibrahim, S.H.; Hirsova, P.; Gores, G.J. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018, 67, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, B.; Marjanovic-Haljilji, M.; Mijac, D.; Lukic, S.; Kapor, S.; Kapor, S.; Starcevic, A.; Popovic, D.; Djokovic, A. Molecular Aspects of MAFLD-New Insights on Pathogenesis and Treatment. Curr. Issues Mol. Biol. 2023, 45, 9132–9148. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and Prevention of Hepatic Steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Cho, Y.E.; Kim, S.J.; Hwang, S. Immune-related pathogenesis and therapeutic strategies of nonalcoholic steatohepatitis. Arch. Pharm. Res. 2022, 45, 229–244. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding roles for SREBP in metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Brown, M.S.; Li, W.P.; Nohturfft, A.; Goldstein, J.L.; Espenshade, P.J. Transport-dependent proteolysis of SREBP: Relocation of site-1 protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell 1999, 99, 703–712. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Horton, J.D.; Shah, N.A.; Warrington, J.A.; Anderson, N.N.; Park, S.W.; Brown, M.S.; Goldstein, J.L. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. USA 2003, 100, 12027–12032. [Google Scholar] [CrossRef]
- Strable, M.S.; Ntambi, J.M. Genetic control of de novo lipogenesis: Role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Hwang, S.; Nguyen, A.D.; Jo, Y.; Engelking, L.J.; Brugarolas, J.; DeBose-Boyd, R.A. Hypoxia-inducible factor 1α activates insulin-induced gene 2 (Insig-2) transcription for degradation of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase in the liver. J. Biol. Chem. 2017, 292, 9382–9393. [Google Scholar] [CrossRef]
- Summons, R.E.; Bradley, A.S.; Jahnke, L.L.; Waldbauer, J.R. Steroids, triterpenoids and molecular oxygen. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 951–968. [Google Scholar] [CrossRef]
- Hwang, S.; Hartman, I.Z.; Calhoun, L.N.; Garland, K.; Young, G.A.; Mitsche, M.A.; McDonald, J.; Xu, F.; Engelking, L.; DeBose-Boyd, R.A. Contribution of Accelerated Degradation to Feedback Regulation of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase and Cholesterol Metabolism in the Liver. J. Biol. Chem. 2016, 291, 13479–13494. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Kim, Y.; Kim, S.J.; Lee, H.; Hwang, S. Overexpression of Interleukin-8 Promotes the Progression of Fatty Liver to Nonalcoholic Steatohepatitis in Mice. Int. J. Mol. Sci. 2023, 24, 15489. [Google Scholar] [CrossRef] [PubMed]
- Hwahng, S.H.; Ki, S.H.; Bae, E.J.; Kim, H.E.; Kim, S.G. Role of adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway in repression of liver X receptor-alpha-dependent lipogenic gene induction and hepatic steatosis by a novel class of dithiolethiones. Hepatology 2009, 49, 1913–1925. [Google Scholar] [CrossRef]
- Bennett, M.K.; Lopez, J.M.; Sanchez, H.B.; Osborne, T.F. Sterol regulation of fatty acid synthase promoter. Coordinate feedback regulation of two major lipid pathways. J. Biol. Chem. 1995, 270, 25578–25583. [Google Scholar] [CrossRef]
- Bené, H.; Lasky, D.; Ntambi, J.M. Cloning and characterization of the human stearoyl-CoA desaturase gene promoter: Transcriptional activation by sterol regulatory element binding protein and repression by polyunsaturated fatty acids and cholesterol. Biochem. Biophys. Res. Commun. 2001, 284, 1194–1198. [Google Scholar] [CrossRef]
- Oh, S.Y.; Park, S.K.; Kim, J.W.; Ahn, Y.H.; Park, S.W.; Kim, K.S. Acetyl-CoA carboxylase beta gene is regulated by sterol regulatory element-binding protein-1 in liver. J. Biol. Chem. 2003, 278, 28410–28417. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Siddiq, A.; Ayoub, I.A.; Chavez, J.C.; Aminova, L.; Shah, S.; LaManna, J.C.; Patton, S.M.; Connor, J.R.; Cherny, R.A.; Volitakis, I.; et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J. Biol. Chem. 2005, 280, 41732–41743. [Google Scholar] [CrossRef]
- Kuiper, C.; Vissers, M.C. Ascorbate as a co-factor for fe- and 2-oxoglutarate dependent dioxygenases: Physiological activity in tumor growth and progression. Front. Oncol. 2014, 4, 359. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Koivunen, P.; Kaelin, W.G., Jr. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat. Rev. Cancer 2020, 20, 710–726. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, D.X.; Shi, X.; Zhang, Q.; Huang, Y.S. Activation of the prolyl-hydroxylase oxygen-sensing signal cascade leads to AMPK activation in cardiomyocytes. J. Cell Mol. Med. 2012, 16, 2049–2059. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar] [CrossRef]
- Yabe, D.; Brown, M.S.; Goldstein, J.L. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 12753–12758. [Google Scholar] [CrossRef]
- Sever, N.; Song, B.L.; Yabe, D.; Goldstein, J.L.; Brown, M.S.; DeBose-Boyd, R.A. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 2003, 278, 52479–52490. [Google Scholar] [CrossRef] [PubMed]
- Engelking, L.J.; Liang, G.; Hammer, R.E.; Takaishi, K.; Kuriyama, H.; Evers, B.M.; Li, W.P.; Horton, J.D.; Goldstein, J.L.; Brown, M.S. Schoenheimer effect explained--feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J. Clin. Investig. 2005, 115, 2489–2498. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Goda, N.; Kanai, M.; Niwa, D.; Osanai, K.; Yamamoto, Y.; Senoo-Matsuda, N.; Johnson, R.S.; Miura, S.; Kabe, Y.; et al. HIF-1α induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J. Hepatol. 2012, 56, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Nath, B.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Catalano, D.; Mandrekar, P.; Szabo, G. Hepatocyte-specific hypoxia-inducible factor-1α is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 2011, 53, 1526–1537. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|---|
| Human | RPLP0(36B4) | CGACCTGGAAGTCCAACTAC | ATCTGCTGCATCTGCTTG |
| Human | SREBP1c | TCGCGGAGCCATGGATT | GGAAGTCACTGTCTTGGTTGTTGA |
| Human | FASN | TCGTGGGCTACAGCATGGT | GCCCTCTGAAGTCGAAGAAGAA |
| Human | ACACA | TTCAGAGGCAGGGTGGGTTA | ACATACTCGTTTGTGTCATAATTTGGT |
| Human | SCD1 | TCACCACATTCTTCATTGATTGC | TTGGAGACTTTCTTCCGGTCAT |
| Human | INSIG1 | CCCAGATTTCCTCTATATTCGTTCTT | CACCCATAGCTAACTGTCGTCCTA |
| Human | INSIG2 | TGTCTCTCACACTGGCTGCACTA | CTCCAAGGCCAAAACCACTTC |
| Human | VEGF | CGCAGCTACTGCCATCCAAT | TGGCTTGAAGATGTACTCGATCTC |
| Human | HIF1A | ATCCATGTGACCATGAGGAAATG | TCGGCTAGTTAGGGTACACTTC |
| Mouse | Rplp0(36B4) | AGATTCGGGATATGCTGTTGGC | TCGGGTCCTAGACCAGTGTTC |
| Mouse | Srebp1c | GGAGCCATGGATTGCACATT | GGCCCGGGAAGTCACTGT |
| Mouse | Fasn | GCTGCGGAAACTTCAGGAAAT | AGAGACGTGTCACTCCTGGACTT |
| Mouse | Acaca | TGGACAGACTGATCGCAGAGAAAG | TGGAGAGCCCCACACACA |
| Mouse | Scd1 | CCGGAGACCCCTTAGATCGA | TAGCCTGTAAAAGATTTCTGCAAACC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, Y.S.; Cho, Y.E.; Kim, Y.; Koh, M.; Hwang, S. Dimethyloxalylglycine Suppresses SREBP1c and Lipogenic Gene Expressions in Hepatocytes Independently of HIF1A. Curr. Issues Mol. Biol. 2024, 46, 2386-2397. https://doi.org/10.3390/cimb46030151
Kwon YS, Cho YE, Kim Y, Koh M, Hwang S. Dimethyloxalylglycine Suppresses SREBP1c and Lipogenic Gene Expressions in Hepatocytes Independently of HIF1A. Current Issues in Molecular Biology. 2024; 46(3):2386-2397. https://doi.org/10.3390/cimb46030151
Chicago/Turabian StyleKwon, Yong Seong, Ye Eun Cho, Yeonsoo Kim, Minseob Koh, and Seonghwan Hwang. 2024. "Dimethyloxalylglycine Suppresses SREBP1c and Lipogenic Gene Expressions in Hepatocytes Independently of HIF1A" Current Issues in Molecular Biology 46, no. 3: 2386-2397. https://doi.org/10.3390/cimb46030151
APA StyleKwon, Y. S., Cho, Y. E., Kim, Y., Koh, M., & Hwang, S. (2024). Dimethyloxalylglycine Suppresses SREBP1c and Lipogenic Gene Expressions in Hepatocytes Independently of HIF1A. Current Issues in Molecular Biology, 46(3), 2386-2397. https://doi.org/10.3390/cimb46030151