
Prenatal Diagnosis by Trio Clinical Exome Sequencing: Single Center Experience

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. CMA and Trio Clinical Exome Sequencing Analysis
3. Results and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wojcik, M.H.; Schwartz, T.S.; Thiele, K.E.; Paterson, H.; Stadelmaier, R.; Mullen, T.E.; VanNoy, G.E.; Genetti, C.A.; Madden, J.A.; Gubbels, C.S.; et al. Infant mortality: The contribution of genetic disorders. J. Perinatol. 2019, 39, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Committee on Genetics and the Society for Maternal-Fetal Medicine. Committee Opinion No.682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obstet. Gynecol. 2016, 128, e262–e268. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Wapner, R. Prenatal diagnosis by chromosomal microarray analysis. Fertil. Steril. 2018, 109, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Best, S.; Wou, K.; Vora, N.; Van Der Veyver, I.B.; Wapner, R.; Chitty, L.S. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat. Diagn. 2018, 38, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Normand, E.A.; Braxton, A.; Nassef, S.; Ward, P.A.; Vetrini, F.; He, W.; Patel, V.; Qu, C.; Westerfield, L.E.; Stover, S.; et al. Clinical exome sequencing for fetuseses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med. 2018, 10, 74. [Google Scholar] [CrossRef]
- Chung, C.C.; Hue, S.P.; Ng, N.Y.; Doong, P.H.; Chu, A.T.; Chung, B.H. Meta-analysis of the diagnostic and clinical utility of exome and genome sequencing in pediatric and adult patients with rare diseases across diverse populations. Anesth. Analg. 2023, 25, 100896. [Google Scholar] [CrossRef] [PubMed]
- Corsten-Janssen, N.; Bouman, K.; Diphoorn, J.C.D.; Scheper, A.J.; Kinds, R.; El Mecky, J.; Breet, H.; Verheij, J.; Suijkerbuijk, R.; Duin, L.K.; et al. A prospective study on rapid exome sequencing as a diagnostic test for multiple congenital anomalies on fetal ultrasound. Prenat. Diagn. 2020, 40, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, H.; Korinth, D.; Ritthaler, M.; Schulte, B.; Battke, F.; Von Kaisenberg, C.; Wustemann, M.; Schulze, B.; Friedrich-Freksa, A.; Pfeiffer, L.; et al. Trio exome sequencing is highly relevant in prenatal diagnostics. Prenat. Diagn. 2022, 42, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Tran Mau-Them, F.; Delanne, J.; Denomme-Pichon, A.S.; Safraou, H.; Bruel, A.L.; Vitobello, A.; Garde, A.; Nambot, S.; Bourgon, N.; Racine, C.; et al. Prenatal diagnosis by trio exome sequencing in fetuseses with ultrasound anomalies: A powerful diagnostic tool. Front. Genet. 2023, 14, 1099995. [Google Scholar] [CrossRef]
- Wou, K.; DeBie, I.; Carroll, J.; Brock, J.-A.; Wilson, R.D. Fetal Exome Sequencing on the Horizon. J. Obstet. Gynaecol. Can. 2019, 41, 64–67. [Google Scholar] [CrossRef]
- Margiotti, K.; Giorlandino, C. Fetal Precision Medicine Achieved with Trio Exome Sequencing Analysis. Clin. Exp. Obstet. Gynecol. 2024, 51, 29. [Google Scholar] [CrossRef]
- Sabbagh, R.; Van Den Veyver, I.B. The current and future impact of genome-wide sequencing on fetal precision medicine. Hum. Genet. 2020, 139, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2020, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Schulz, M.H.; Krawitz, P.; Bauer, S.; Dölken, S.; Ott, C.E.; Mundlos, C.; Horn, D.; Mundlos, S.; Robinson, P.N. Clinical Diagnostics in Human Genetics with Semantic Similarity Searches in Ontologies. Am. J. Hum. Genet. 2009, 85, 457–464. [Google Scholar] [CrossRef]
- Spaggiari, E.; Faure, G.; Rousseau, V.; Sonigo, P.; Millischer-Bellaiche, A.; Kermorvant-Duchemin, E.; Muller, F.; Czerkiewicz, I.; Ville, Y.; Salomon, L.J. Performance of prenatal diagnosis in esophageal atresia. Prenat. Diagn. 2015, 35, 888–893. [Google Scholar] [CrossRef]
- Boito, S.; Crovetto, F.; Ischia, B.; Crippa, B.L.; Fabietti, I.; Bedeschi, M.F.; Lalatta, F.; Colombo, L.; Mosca, F.; Fedele, L.; et al. Prenatal ultrasound factors and genetic disorders in pregnancies complicated by polyhydramnios. Prenat. Diagn. 2016, 36, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Dorleijn, D.M.J.; Cohen-Overbeek, T.E.; Groenendaal, F.; Bruinse, H.W.; Stoutenbeek, P. Idiopathic polyhydramnios and postnatal findings. J. Matern. Neonatal Med. 2009, 22, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Estep, A.L.; Tidyman, W.E.; Teitell, M.A.; Cotter, P.D.; Rauen, K.A. HRAS mutations in Costello syndrome: Detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am. J. Med. Genet. Part A 2005, 140A, 8–16. [Google Scholar] [CrossRef]
- Musante, L.; Kehl, H.G.; Majewski, F.; Meinecke, P.; Schweiger, S.; Gillessen-Kaesbach, G.; Wieczorek, D.; Hinkel, G.K.; Tinschert, S.; Hoeltzenbein, M.; et al. Spectrum of mutations in PTPN11 and genotype-phenotype correlation in 96 patients with Noonan syndrome and five patients with cardio-facio-cutaneous syndrome. Eur. J. Hum. Genet. 2003, 11, 201–206. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Mangels, R.; Blumenfeld, Y.J.; Homeyer, M.; Mrazek-Pugh, B.; Hintz, S.R.; Hudgins, L. RASopathies: A significant cause of polyhydramnios? Prenat. Diagn. 2021, 41, 362–367. [Google Scholar] [CrossRef]
- Scott, A.; Di Giosaffatte, N.; Pinna, V.; Daniele, P.; Corno, S.; D’ambrosio, V.; Andreucci, E.; Marozza, A.; Sirchia, F.; Tortora, G.; et al. When to test fetuseses for RASopathies? Proposition from a systematic analysis of 352 multicenter cases and a postnatal cohort. Genet. Med. 2021, 23, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.J.; Norton, M.E.; Shaw, G.M.; Flessel, M.C.; Goldman, S.; Currier, R.J.; Jelliffe-Pawlowski, L.L. Risk of selected structural abnormalities in infants after increased nuchal translucency measurement. Am. J. Obstet. Gynecol. 2014, 211, 675.e1–675.e19. [Google Scholar] [CrossRef]
- Grande, M.; Jansen, F.A.; Blumenfeld, Y.J.; Fisher, A.; Odibo, A.O.; Haak, M.C.; Borrell, A. Genomic microarray in fetuseses with increased nuchal translucency and normal karyotype: A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2015, 46, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Wu, H.; She, L.; Zhang, B.; He, Y.; Luo, D.; Wang, H.; Zheng, Z. Analysis of copy number variation by sequencing in fetuseses with nuchal translucency thickening. J. Clin. Lab. Anal. 2020, 34, e23347. [Google Scholar] [CrossRef] [PubMed]
- Matyasova, M.; Dobsakova, Z.; Hiemerova, M.; Kadlecova, J.; Nikulenkov Grochova, D.; Popelinska, E.; Svobodova, E.; Vlasin, P. Prenatal diagnosis of Noonan syndrome in fetuseses with increased nuchal translucency and a normal karyotype. Ceska Gynekol. 2019, 84, 195–200. [Google Scholar]
- Bukowska-Olech, E.; Trzebiatowska, W.; Czech, W.; Drzymala, O.; Frak, P.; Klarowski, F.; Klusek, P.; Szwajkowska, A.; Jamsheer, A. Hereditary Multiple Exostoses-A Review of the Molecular Background, Diagnostics, and Potential Therapeutic Strategies. Front. Genet. 2021, 12, 759129. [Google Scholar] [CrossRef] [PubMed]
- Stieber, J.R.; Dormans, J.P. Manifestations of Hereditary Multiple Exostoses. J. Am. Acad. Orthop. Surg. 2005, 13, 110–120. [Google Scholar] [CrossRef]
- Smith, D.W.; Lemli, L.; Opitz, J.M. A Newly Recognized Syndrome of Multiple Congenital Anomalies. J. Pediatr. 1964, 64, 210–217. [Google Scholar] [CrossRef]
- FitzPatrick, D.R. Zellweger syndrome and associated phenotypes. J. Med. Genet. 1996, 33, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Bachmann-Gagescu, R.; E Ishak, G.; Dempsey, J.C.; Adkins, J.; O’Day, D.; Phelps, I.G.; Gunay-Aygun, M.; Kline, A.D.; Szczaluba, K.; Martorell, L.; et al. Genotype–phenotype correlation in CC2D2A-related Joubert syndrome reveals an association with ventriculomegaly and seizures. J. Med. Genet. 2012, 49, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, L.; Dubourg, C.; Pasquier, L.; Le Duff, F.; Blayau, M.; Durou, M.R.; De La Pintiere, A.T.; Aguilella, C.; David, V.; Odent, S. Phenotypic and molecular variability of the holoprosencephalic spectrum. Am. J. Med. Genet. Part A 2004, 129A, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Nawaz, A.; Hamid, M.; Ullah, W.; Khan, I.N.; Afshan, M.; Rehman, A.; Nawaz, H.; Halswick, J.; Rehman, S.U.; et al. Mutation screening of multiple Pakistani MCPH families revealed novel and recurrent protein-truncating mutations of ASPM. Biotechnol. Appl. Biochem. 2022, 69, 2296–2303. [Google Scholar] [CrossRef]
- Clementi, M.; Tenconi, R.; Bianchi, F.; Stoll, C. Evaluation of prenatal diagnosis of cleft lip with or without cleft palate and cleft palate by ultrasound: Experience from 20 European registries. Prenat. Diagn. 2000, 20, 870–875. [Google Scholar] [CrossRef]
- Jain, M.; De Jesus, O. Krabbe Disease; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Gabrielli, S.; Piva, M.; Ghi, T.; Perolo, A.; De Santis, M.S.N.; Bevini, M.; Bonasoni, P.; Santini, D.; Rizzo, N.; Pilu, G. Bilateral cleft lip and palate without premaxillary protrusion is associated with lethal aneuploidies. Ultrasound Obstet. Gynecol. 2009, 34, 416–418. [Google Scholar] [CrossRef]


{kind=link}
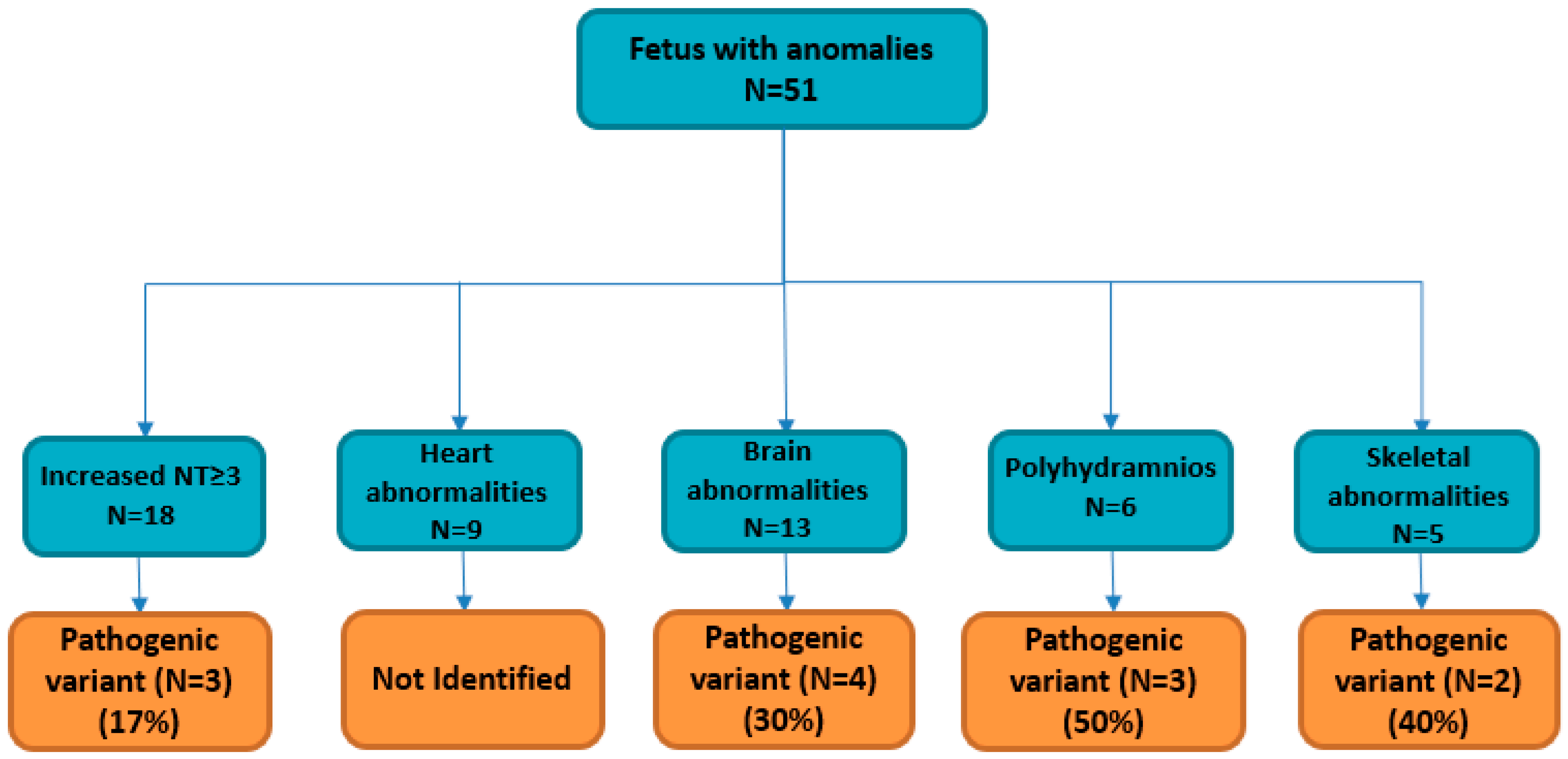
| Primary Ultrasound Anomaly | Total Cases | Solved Cases (Diagnostic Yield) |
|---|---|---|
| Increased NT ≥ 3 mm * | 18 | 3 (17%) |
| Heart abnormalities | 9 | ND |
| Brain abnormalities | 13 | 4 (30%) |
| Polyhydramnios | 6 | 3 (50%) |
| Skeletal abnormalities | 5 | 2 (40%) |
| Fetal sex | ||
| Male | 23 | |
| Famele | 28 |
| Fetuses | Gene | Transcript Change | Protein Change | Primary Ultrasound Signs | Secondary Ultrasound Signs | Fetal Phenotype (HP **) | Zigosity | Transmission | ClinVar/HGMD * | OMIM Gene | Disease | OMIM Disease | Inheritance |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GALC | c.379C>T; c.863G>A | p.Arg127Ter; p.Trp288Ter | Bilateral cleft lip/palate | NA | HP:0002744 | Compound heterozygosity | Biparental | Path. | 606890 | Krabbe disease | 245200 | AR |
| 2 | SIX3 | c.385G>T | p.Glu129Ter | Holoprosencephaly | NA | HP:0001360 | Heterozygosity | De Novo | Path. | 603714 | Holoprosencephaly 2 | 157170 | AD |
| 3 | EXT2 | c.429C>G | p.Y143Ter | Increased NT = 3,0 | NA | HP:0010880 | Heterozygosity | De Novo | Path. | 608210 | Exostoses, multiple, type 2 | 133701 | AD |
| 4 | EXT1 | c.1818G>A | p.Trp606Ter | Increased NT = 3,0 | NA | HP:0010880 | Heterozygosity | Paternal | Path. | 608177 | Exostoses, multiple, type 1 | 133700 | AD |
| 5 | DHCR7 | c.453G>A | p.W151X | Increased NT = 3,5 | Renal anomalies | HP:0010880; HP:0000077 | Homozygous | Biparental | Path. | 602858 | Smith–Lemli–Opitz syndrome | 270400 | AR |
| 6 | ASPM | c.3055C>T | p.Arg1019Ter | Microcephaly | NA | HP:0000252 | Homozygous | Biparental | Path. | 605481 | Microcephaly 5, primary, autosomal recessive | 608716 | AR |
| 7 | HRAS | c.37G>T | p.Gly13Cys | Polyhydramnios | Pleural effusion | HP:0001561; HP:0002202 | Heterozygosity | De Novo | Path. | 190020 | Costello syndrome | 218040 | AD |
| 8 | PTPN11 | c.174C>G | p.Asn58Lys | Polyhydramnios | NA | HP:0001562 | Heterozygosity | De Novo | Path. | 176876 | Noonan syndrome 1 | 163950 | AD |
| 9 | PTPN11 | c.923A>G | p.Asn308Ser | Polyhydramnios | Renal anomalies | HP:0001563; HP:0000077 | Heterozygosity | De Novo | Path. | 176876 | Noonan syndrome 1 | 163950 | AD |
| 10 | COL1A1 | c.2684dupC | p.Gly896fs | Short fetal femur length | NA | HP:0011428 | Heterozygosity | De Novo | Path. | 120150 | Osteogenesis imperfecta, type I | 166200 | AD |
| 11 | PEX1 | c.2528G>A | p.Gly843Asp | Ventriculomegaly | NA | HP:0002119 | Homozygous | Biparental | Path. | 602136 | Heimler syndrome 1 | 234580 | AR |
| 12 | CC2D2A | c.3850C>T | p.Arg1284Cys | Ventriculomegaly | NA | HP:0002119 | Homozygous | Biparental | Path. | 612013 | Meckel syndrome 6 | 612284 | AR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margiotti, K.; Fabiani, M.; Cima, A.; Libotte, F.; Mesoraca, A.; Giorlandino, C. Prenatal Diagnosis by Trio Clinical Exome Sequencing: Single Center Experience. Curr. Issues Mol. Biol. 2024, 46, 3209-3217. https://doi.org/10.3390/cimb46040201
Margiotti K, Fabiani M, Cima A, Libotte F, Mesoraca A, Giorlandino C. Prenatal Diagnosis by Trio Clinical Exome Sequencing: Single Center Experience. Current Issues in Molecular Biology. 2024; 46(4):3209-3217. https://doi.org/10.3390/cimb46040201
Chicago/Turabian StyleMargiotti, Katia, Marco Fabiani, Antonella Cima, Francesco Libotte, Alvaro Mesoraca, and Claudio Giorlandino. 2024. "Prenatal Diagnosis by Trio Clinical Exome Sequencing: Single Center Experience" Current Issues in Molecular Biology 46, no. 4: 3209-3217. https://doi.org/10.3390/cimb46040201
APA StyleMargiotti, K., Fabiani, M., Cima, A., Libotte, F., Mesoraca, A., & Giorlandino, C. (2024). Prenatal Diagnosis by Trio Clinical Exome Sequencing: Single Center Experience. Current Issues in Molecular Biology, 46(4), 3209-3217. https://doi.org/10.3390/cimb46040201