
The Molecular Mechanisms Underlying the Systemic Effects Mediated by Parathormone in the Context of Chronic Kidney Disease

 , , , , ,
, , , , ,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Parathyroid Hormone Physiological Actions
4. Chronic Kidney Disease Rapidly Leads to Mineral and Bone Disorder
4.1. Classical View of CKD-MBD
4.2. Updated “Trade-Off Hypothesis”
4.2.1. FGF-23, the Missing Piece from the Puzzle
4.2.2. Uremic Conditions Diminish the Potential Benefit of Increased Circulating FGF-23
4.2.3. Klotho—An Aging-Related Molecule
4.2.4. Vitamin D Encloses the Hormonal Triangle
5. Biochemical Diagnosis of CKD-MBD: Focus on PTH
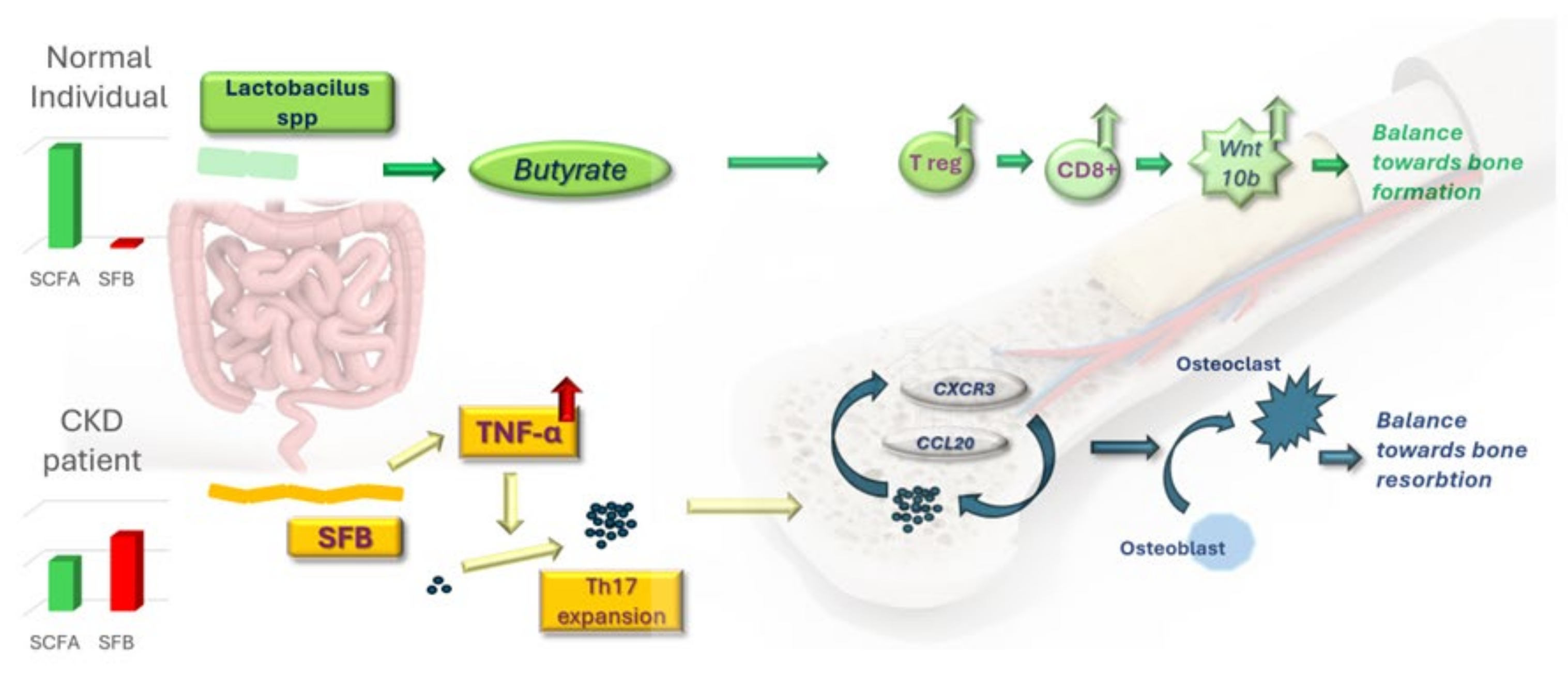
6. Intestinal Microbiota in Mediating PTH Actions on Bone Turnover
6.1. Intestinal Microbiota Acts as Vehicle in Mediating Bone Loss in Hyperparathyroidism
6.2. Short-Chain Fatty Acids Are Fundamental in Mediating PTH-Dependent Bone Formation
6.3. Damaged Gut Microbiota Takes a Toll on Bone Homeostasis: Can Probiotics Save It?
7. Secondary Hyperparathyroidism and Bone Homeostasis
7.1. PTH Signaling Exhibits a Dual Behaviour in Bone Tissue
7.2. Recent Advances in PTH Signaling Mechanisms with Respect to Bone
7.2.1. Dkk1 as an Intermediate Marker of PTH Signaling
7.2.2. Sclerostin Regulation—Downstream Cascades in PTH Signaling May Be More Powerful Than PTH Itself
7.2.3. PTH Is the Central Element in Two Progressively Aggravating Entities with Systemic Reverb: Renal Osteodystrophy and Cardiovascular Disease
7.2.4. Fracture Risk Is an Indirect Measure of Cortical Bone Mass and Mineralization Efficiency
8. Parathormone Role in Determining Cardiovascular Disease: Friend or Foe?
8.1. Physiological Benefits of PTHR1 Activation—Antiapoptotic Behaviour and Systemic Vasodilatory Potential
8.2. Paradoxically, Hyperparathyroidism Does Not Manifest the Benefits of PTH Signaling
8.3. The Cardiovascular “Snowball Effect” of Secondary Hyperparathyroidism
8.3.1. Aldosterone/PTH Vicious Synergic Coupling
8.3.2. High Levels of PTH Allow for Increased Renin Plasma Activity and Secretion of Angiotensin II: A Closed Loop
8.4. Vascular Calcification—The Implication of PTH
8.5. The Key to Assessing Vascular PTH Axis Status Could Lie in the Endothelial Involvement in PTH Signaling
9. Management of Biochemical Abnormalities in CKD-MBD Focusing on PTH Level Changes
9.1. Limitation of Phosphate Retention Attenuates PTH Levels
9.2. Manipulation of Calcium-Sensing Receptor Yields Beneficial Effects on Progression of Mineral and Bone Disorder
9.3. Vitamin D Supplementation Yields Disparate Results Depending on the Analogue
9.4. Anabolic Agents
9.5. The Lower the PTH Levels, the Better? Not in Every Circumstance
10. NHERF1—In the Shadow of PTH Signaling
10.1. The Physiological Coupling of Scaffolding Protein NHERF1 and PTHR1
10.2. Reduced Expression of NHERF1 Leads the Way for Various Renal Alterations
11. Discussions
12. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Saran, R.; Robinson, B.; Abbott, K.C.; Agodoa, L.Y.C.; Bragg-Gresham, J.; Balkrishnan, R.; Bhave, N.; Dietrich, X.; Ding, Z.; Eggers, P.W.; et al. US Renal Data System 2018 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2019, 73, A7–A8. [Google Scholar] [CrossRef] [PubMed]
- Schrauben, S.J.; Chen, H.Y.; Lin, E.; Jepson, C.; Yang, W.; Scialla, J.J.; Fischer, M.J.; Lash, J.P.; Fink, J.C.; Hamm, L.L.; et al. Hospitalizations among adults with chronic kidney disease in the United States: A cohort study. PLoS Med. 2020, 17, e1003470. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Mone, P.; Jankauskas, S.S.; Gambardella, J.; Santulli, G. Chronic kidney disease: Definition, updated epidemiology, staging, and mechanisms of increased cardiovascular risk. J. Clin. Hypertens. 2021, 23, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Shlipak, M.G.; Tummalapalli, S.L.; Boulware, L.E.; Grams, M.E.; Ix, J.H.; Jha, V.; Kengne, A.P.; Madero, M.; Mihaylova, B.; Tangri, N.; et al. The case for early identification and intervention of chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2021, 99, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Wouters, O.J.; O’Donoghue, D.J.; Ritchie, J.; Kanavos, P.G.; Narva, A.S. Early chronic kidney disease: Diagnosis, management and models of care. Nat. Rev. Nephrol. 2015, 11, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Recio-Mayoral, A.; Banerjee, D.; Streather, C.; Kaski, J.C. Endothelial dysfunction, inflammation and atherosclerosis in chronic kidney disease—A cross-sectional study of predialysis, dialysis and kidney-transplantation patients. Atherosclerosis 2011, 216, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Schiffrin, E.L.; Lipman, M.L.; Mann, J.F. Chronic kidney disease: Effects on the cardiovascular system. Circulation 2007, 116, 85–97. [Google Scholar] [CrossRef] [PubMed]
- D’Arrigo, G.; Mallamaci, F.; Pizzini, P.; Leonardis, D.; Tripepi, G.; Zoccali, C. CKD-MBD biomarkers and CKD progression: An analysis by the joint model. Nephrol. Dial. Transpl. 2023, 38, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Goltzman, D. Physiology of Parathyroid Hormone. Endocrinol. Metab. Clin. N. Am. 2018, 47, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9, S1. [Google Scholar] [CrossRef] [PubMed]
- Christakos, S.; Li, S.; De La Cruz, J.; Shroyer, N.F.; Criss, Z.K.; Verzi, M.P.; Fleet, J.C. Vitamin D and the intestine: Review and update. J. Steroid. Biochem. Mol. Biol. 2020, 196, 105501. [Google Scholar] [CrossRef] [PubMed]
- Topala, C.N.; Schoeber, J.P.; Searchfield, L.E.; Riccardi, D.; Hoenderop, J.G.; Bindels, R.J. Activation of the Ca2+-sensing receptor stimulates the activity of the epithelial Ca2+ channel TRPV5. Cell Calcium. 2009, 45, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Déliot, N.; Hernando, N.; Horst-Liu, Z.; Gisler, S.M.; Capuano, P.; Wagner, C.A.; Bacic, D.; O’Brien, S.; Biber, J.; Murer, H. Parathyroid hormone treatment induces dissociation of type IIa Na+-Pi cotransporter-Na+/H+ exchanger regulatory factor-1 complexes. Am. J. Physiol. Cell Physiol. 2005, 289, C159–C167. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef]
- Kidney Disease Improving Global Outcomes Work Group. Chapter 4: Other complications of CKD: CVD, medication dosage, patient safety, infections, hospitalizations, and caveats for investigating complications of CKD. Kidney Int. Suppl. 2013, 3, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2017, 7, 1–59. [Google Scholar] [CrossRef] [PubMed]
- Slatopolsky, E. The intact nephron hypothesis: The concept and its implications for phosphate management in CKD-related mineral and bone disorder. Kidney Int. Suppl. 2011, 79, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Bricker, N.S.; Morrin, P.A.; Kime, S.W., Jr. The pathologic physiology of chronic Bright’s disease. An exposition of the “intact nephron hypothesis”. J. Am. Soc. Nephrol. 1997, 8, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Bricker, N.S. On the pathogenesis of the uremic state. An exposition of the “trade-off hypothesis”. N. Engl. J. Med. 1972, 286, 1093–1099. [Google Scholar] [CrossRef]
- Gutiérrez, O.M. Fibroblast growth factor 23 and disordered vitamin D metabolism in chronic kidney disease: Updating the “trade-off” hypothesis. Clin. J. Am. Soc. Nephrol. 2010, 5, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Moranne, O.; Froissart, M.; Rossert, J.; Gauci, C.; Boffa, J.J.; Haymann, J.P.; M’Rad, M.B.; Jacquot, C.; Houillier, P.; Stengel, B.; et al. Timing of onset of CKD-related metabolic complications. J. Am. Soc. Nephrol. 2009, 20, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498. [Google Scholar] [CrossRef] [PubMed]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Overview of the FGF23-Klotho axis. Pediatr. Nephrol. 2010, 25, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Moe, O.W. Role of αKlotho and FGF23 in regulation of type II Na-dependent phosphate co-transporters. Pflugers Arch. 2019, 471, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutierrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Ito, M.; Kuwahata, M.; Kato, S.; Segawa, H. Inhibition of intestinal sodium-dependent inorganic phosphate transport by fibroblast growth factor 23. Ther. Apher. Dial. 2005, 9, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Canalejo, R.; Canalejo, A.; Martinez-Moreno, J.M.; Rdoriguez-Ortiz, M.E.; Estapa, J.C.; Mendoza, F.J.; Munoz-Castaneda, J.R.; Shalhoub, V.; Almaden, Y.; Rodriguez, M. FGF23 fails to inhibit uremic parathyroid glands. J. Am. Soc. Nephrol. 2010, 21, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Renal and extrarenal actions of Klotho. Semin. Nephrol. 2013, 33, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.; Locatelli, F.; Rodriguez, M. Secondary hyperparathyroidism: Pathogenesis, disease progression, and therapeutic options. Clin. J. Am. Soc. Nephrol. 2011, 6, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Renal Physiol. 2005, 289, F8–F28. [Google Scholar] [CrossRef] [PubMed]
- Girgis, C.M.; Clifton-Bligh, R.J.; Hamrick, M.W.; Holick, M.F.; Gunton, J.E. The roles of vitamin D in skeletal muscle: Form, function, and metabolism. Endocr. Rev. 2013, 34, 33–83. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.G.; Hsu, S.; de Boer, I.H. Vitamin D supplementation in people with chronic kidney disease. Kidney Int. 2023, 104, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Napoletano, A.; Provenzano, M.; Garofalo, C.; Bini, C.; Comai, G.; La Manna, G. Mineral Bone Disorders in Kidney Disease Patients: The Ever-Current Topic. Int. J. Mol. Sci. 2022, 23, 12223. [Google Scholar] [CrossRef] [PubMed]
- Cavalier, E.; Lukas, P.; Bottani, M.; Aarsand, A.K.; Ceriotti, F.; Coskun, A.; Diaz-Garzon, J.; Fernandez-Calle, P.; Guerra, E.; Locatelli, M.; et al. European Biological Variation Study (EuBIVAS): Within- and between-subject biological variation estimates of β-isomerized C-terminal telopeptide of type I collagen (β-CTX), N-terminal propeptide of type I collagen (PINP), osteocalcin, intact fibroblast growth factor 23 and uncarboxylated-unphosphorylated matrix-Gla protein-a cooperation between the EFLM Working Group on Biological Variation and the International Osteoporosis Foundation-International Federation of Clinical Chemistry Committee on Bone Metabolism. Osteoporos. Int. 2020, 31, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Cavalier, E.; Delanaye, P. Variability of New Bone Mineral Metabolism Markers in Patients Treated with Maintenance Hemodialysis: Implications for Clinical Decision Making. Am. J. Kidney Dis. 2013, 61, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Heijboer, A.C.; Cavalier, E. The Measurement and Interpretation of Fibroblast Growth Factor 23 (FGF23) Concentrations. Calcif. Tissue Int. 2023, 112, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Amrein, K.; Scherkl, M.; Hoffmann, M.; Neuwersch-Sommeregger, S.; Kostenberger, M.; Berisha, A.T.; Martucci, G.; Pilz, S.; Malle, O. Vitamin D deficiency 2.0: An update on the current status worldwide. Eur. J. Clin. Nutr. 2020, 74, 1498–1513. [Google Scholar] [CrossRef] [PubMed]
- Souberbielle, J.C.; Roth, H.; Fouque, D.P. Parathyroid hormone measurement in CKD. Kidney Int. 2010, 77, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Scheibel, S.; D’Amour, P.; John, M.R.; Rao, S.D.; Schmidt-Gayk, H.; Cantor, T.L. Development of a novel immunoradiometric assay exclusively for biologically active whole parathyroid hormone 1-84: Implications for improvement of accurate assessment of parathyroid function. J. Bone Miner. Res. 2001, 16, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Souberbielle, J.C.; Boutten, A.; Carlier, M.C.; Chevenne, D.; Coumaros, G.; Lawson-Body, E.; Massart, C.; Monge, M.; Myara, J.; Parent, X.; et al. Inter-method variability in PTH measurement: Implication for the care of CKD patients. Kidney Int. 2006, 70, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Monier-Faugere, M.C.; Geng, Z.; Mawad, H.; Friedler, R.M.; Gao, P.; Cantor, T.L.; Malluche, H.H. Improved assessment of bone turnover by the PTH-(1-84)/large C-PTH fragments ratio in ESRD patients. Kidney Int. 2001, 60, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Sprague, S.M.; Moe, S.M. The case for routine parathyroid hormone monitoring. Clin. J. Am. Soc. Nephrol. 2013, 8, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Rudser, K.D.; de Boer, I.H.; Dooley, A.; Young, B.; Kestenbaum, B. Fracture risk after parathyroidectomy among chronic hemodialysis patients. J. Am. Soc. Nephrol. 2007, 18, 2401–2407. [Google Scholar] [CrossRef] [PubMed]
- Hagström, E.; Hellman, P.; Larsson, T.E.; Ingelsson, E.; Berglund, L.; Sundstrom, J.; Melhus, H.; Held, C.; Lind, L.; Michaelsson, K.; et al. Plasma parathyroid hormone and the risk of cardiovascular mortality in the community. Circulation 2009, 119, 2765–2771. [Google Scholar] [CrossRef] [PubMed]
- Cannata-Andía, J.B.; Carrillo-López, N.; Rodriguez-García, M.; Torregrosa, J.V. Mineral and Bone Disorders in Chronic Kidney Disease. In Management of Chronic Kidney Disease, 1st ed.; Arici, M., Ed.; Springer: Cham, Switzerland, 2023; pp. 239–256. [Google Scholar] [CrossRef]
- Li, J.Y.; Yu, M.; Pal, S.; Tyagi, A.M.; Dar, H.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; Pacifici, R. Parathyroid hormone-dependent bone formation requires butyrate production by intestinal microbiota. J. Clin. Investig. 2020, 130, 1767–1781. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A.; Drueke, T.B. Gut microbiota orchestrates PTH action in bone: Role of butyrate and T cells. Kidney Int. 2020, 98, 269–272. [Google Scholar] [CrossRef]
- Yu, M.; Tyagi, A.M.; Li, J.Y.; Adams, J.; Denning, T.L.; Weitzmann, M.N.; Jones, R.M.; Pacifici, R. PTH induces bone loss via microbial-dependent expansion of intestinal TNF+ T cells and Th17 cells. Nat. Commun. 2020, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhou, T.; He, Y.; Xie, Y.; Xu, Y.; Huang, W. The role and mechanism of butyrate in the prevention and treatment of diabetic kidney disease. Front. Microbiol. 2022, 13, 961536. [Google Scholar] [CrossRef]
- Gonzalez, A.; Krieg, R.; Massey, H.D.; Carl, D.; Ghosh, S.; Gehr, T.W.B.; Ghosh, S.S. Sodium butyrate ameliorates insulin resistance and renal failure in CKD rats by modulating intestinal permeability and mucin expression. Nephrol. Dial. Transplant. 2019, 34, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Amiri, P.; Hosseini, S.A.; Ghaffari, S.; Tutunchi, H.; Ghaffari, S.; Mosharkesh, E.; Asghari, S.; Roshanravan, N. Role of Butyrate, a Gut Microbiota Derived Metabolite, in Cardiovascular Diseases: A comprehensive narrative review. Front. Pharmacol. 2022, 12, 837509. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Chi, H. Metabolic control of regulatory T cell development and function. Trends Immunol. 2015, 36, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Bedi, B.; Li, J.Y.; Tawfeek, H.; Baek, K.H.; Adams, J.; Vangara, S.S.; Chang, M.K.; Kneissel, M.; Weitzmann, M.N.; Pacifici, R. Silencing of parathyroid hormone (PTH) receptor 1 in T cells blunts the bone anabolic activity of PTH. Proc. Natl. Acad. Sci. USA 2012, 109, E725–E733. [Google Scholar] [CrossRef] [PubMed]
- Bellido, T.; Ali, A.A.; Gubrij, I.; Plotkin, L.I.; Fu, Q.; O’Brien, C.A.; Manolagas, S.C.; Jilka, R.L. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: A novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 2005, 146, 4577–4583. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Tu, X.; Pacheco-Costa, R.; McAndrews, K.; Edwards, R.; Pellegrini, G.G.; Kuhlenschmidt, K.; Olivos, N.; Robling, A.; Peacock, M.; et al. Control of Bone Anabolism in Response to Mechanical Loading and PTH by Distinct Mechanisms Downstream of the PTH Receptor. J. Bone Miner. Res. 2017, 32, 522–535. [Google Scholar] [CrossRef]
- Kondo, M.; Torisu, T.; Nagasue, T.; Shibata, H.; Umeno, J.; Kawasaki, K.; Fujioka, S.; Matsuno, Y.; Moryiama, T.; Kitazono, T. Duodenal microbiome in chronic kidney disease. Clin. Exp. Nephrol. 2023, 28, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lv, D.; Jiang, S.; Jiang, J.; Liang, M.; Hou, F.; Chen, Y. Quantitative reduction in short-chain fatty acids, especially butyrate, contributes to the progression of chronic kidney disease. Clin. Sci. 2019, 133, 1857–1870. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Jose, A.; Alonzo-Palma, N.; Malik, T.; Shankaranarayanan, D.; Regunathan-Shenk, R.; Raj, D.S. Butyrate producing microbiota are reduced in chronic kidney diseases. Sci. Rep. 2021, 11, 23530. [Google Scholar] [CrossRef] [PubMed]
- Terpstra, M.L.; Sinnige, M.J.; Hugenholtz, F.; Peters-Sengers, H.; Remmerswaal, E.B.; Geerlings, S.E.; Bemelman, F.J. Butyrate production in patients with end-stage renal disease. Int. J. Nephrol. Renovasc. Dis. 2019, 12, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.M.; Yu, M.; Darby, T.M.; Vaccaro, C.; Li, J.Y.; Owens, J.A.; Hsu, E.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; et al. The Microbial Metabolite Butyrate Stimulates Bone Formation via T Regulatory Cell-Mediated Regulation of WNT10B Expression. Immunity 2018, 49, 1116–1131. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.L.; Irwin, R.; Bierhalter, H.; Schepper, J.; Britton, R.A.; Parameswaran, N.; McCabe, L.R. Lactobacillus reuteri 6475 Increases Bone Density in Intact Females Only under an Inflammatory Setting. PLoS ONE 2016, 11, e0153180. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, K.; Lee, Y.; Chen, M. Preventive Effects of Lactobacillus Mixture against Chronic Kidney Disease Progression through Enhancement of Beneficial Bacteria and Downregulation of Gut-Derived Uremic Toxins. J. Agric. Food Chem. 2021, 69, 7353–7366. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, H.; Valkenburg, S.; Madella, A.M.; Garssen, J.; van Bergenhenegouwen, J.; Overbeek, S.A.; Huys, G.R.B.; Raes, J.; Glorieux, G. Gut microbiome studies in CKD: Opportunities, pitfalls and therapeutic potential. Nat. Rev. Nephrol. 2023, 19, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.; Omata, Y.; Hofmann, J.; Bottcher, M.; Iljazovic, A.; Sarter, K.; Albrecht, O.; Schulz, O.; Krishnacoumar, B.; Kronke, G.; et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat. Commun. 2018, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Pazianas, M.; Miller, P.D. The rationale for intermittent administration of PTH in the management of mineral and bone disorder of chronic kidney disease. J. Nephrol. 2023, 1–6. [Google Scholar] [CrossRef]
- Sebastian, E.M.; Suva, L.J.; Friedman, P.A. Differential effects of intermittent PTH (1-34) and PTH (7-34) on bone microarchitecture and aortic calcification in experimental renal failure. Bone 2008, 43, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, M.; Cheung, J.; Moore, A.E.; Frost, M.L.; Fraser, W.D.; Fogelman, I.; Hampson, G. Circulating fibroblast growth factor-23 increases following intermittent parathyroid hormone (1–34) in postmenopausal osteoporosis: Association with biomarker of bone formation. Calcif. Tissue Int. 2010, 87, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, H.; Qin, L.; Tamasi, J.; Bergenstock, M.; Shapses, S.; Feyen, J.H.M.; Notterman, D.A.; Partridge, N.C. Determination of dual effects of parathyroid hormone on skeletal gene expression in vivo by microarray and network analysis. J. Biol. Chem. 2007, 282, 33086–33097. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lee, S.K.; Adams, D.J.; Gronowicz, G.A.; Kream, B.E. CREM deficiency in mice alters the response of bone to intermittent parathyroid hormone treatment. Bone 2007, 40, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Bellido, T.; Ali, A.A.; Plotkin, L.I.; Fu, Q.; Gubrij, I.; Roberson, P.K.; Weinstein, R.S.; O’Brien, C.A.; Manolagas, S.C.; Jilka, R.L. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J. Biol. Chem. 2003, 278, 50259–50272. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Ren, S.; Duffield, J.S. Wnt signalling in kidney diseases: Dual roles in renal injury and repair. J. Pathol. 2013, 229, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Pietila, I.; Ellwanger, K.; Railo, A.; Jokela, T.; Barco Barrantes, I.; Shan, J.; Niehrs, C.; Vainio, S.J. Secreted Wnt antagonist Dickkopf-1 controls kidney papilla development coordinated by Wnt-7b signalling. Dev. Biol. 2011, 353, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Ginsberg, C.; Seifert, M.; Agapova, O.; Sugatani, T.; Register, T.C.; Freedman, B.I.; Monier-Faugere, M.C.; Malluche, H.; Hruska, K.A. CKD-induced wingless/integration1 inhibitors and phosphorus cause the CKD-mineral and bone disorder. J. Am. Soc. Nephrol. 2014, 25, 1760–1773. [Google Scholar] [CrossRef] [PubMed]
- Forster, C.M.; White, C.A.; Turner, M.E.; Norman, P.A.; Ward, E.C.; Hopman, W.M.; Adams, M.A.; Holden, R.M. Circulating Levels of Dickkopf-Related Protein 1 Decrease as Measured GFR Declines and Are Associated with PTH Levels. Am. J. Nephrol. 2020, 51, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, M.; Yang, D.; Bouxsein, M.L.; Saito, H.; Galvin, R.J.S.; Kuhtoss, S.A.; Thomas, C.C.; Schipani, E.; Baron, R.; et al. Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated stromal cell response and new bone formation. Cell Metab. 2010, 11, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.S.; Murray, D.W.; Hurson, C.J.; O’Brien, J.; Doran, P.P.; O’Byrne, J.M. The role of Dkk1 in bone mass regulation: Correlating serum Dkk1 expression with bone mineral density. J. Orthop. Res. 2011, 29, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Gifre, L.; Ruiz-Gaspà, S.; Monegal, A.; Nomdedeu, B.; Filella, X.; Guanabens, N.; Peris, P. Effect of glucocorticoid treatment on Wnt signalling antagonists (sclerostin and Dkk-1) and their relationship with bone turnover. Bone 2013, 57, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, V.M.; Verhulst, A.; Babler, A.; D’Haese, P.C.; Evenepoel, P.; Kaesler, N. Sclerostin in chronic kidney disease-mineral bone disorder think first before you block it! Nephrol. Dial. Transplant. 2019, 34, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Thambiah, S.; Roplekar, R.; Manghat, P.; Fogelman, I.; Fraser, W.D.; Goldsmith, D.; Hampson, G. Circulating sclerostin and Dickkopf-1 (DKK1) in predialysis chronic kidney disease (CKD): Relationship with bone density and arterial stiffness. Calcif. Tissue Int. 2012, 90, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.G.; Massy, Z.A.; Brandenburg, V.M.; Mazafero, S.; Cozzolino, M.; Urena-Torres, P.; Bover, J.; Goldsmith, D.; CKD-MBD Working Group of ERA-EDTA. Bone: A new endocrine organ at the heart of chronic kidney disease and mineral and bone disorders. Lancet Diabetes Endocrinol. 2014, 2, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Ginsberg, C.; Sugatani, T.; Monier-Faugere, M.C.; Malluche, H.; Hruska, K.A. Early chronic kidney disease-mineral bone disorder stimulates vascular calcification. Kidney Int. 2014, 85, 142–150. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Plotkin, L.I.; Galli, C.; Goellner, J.J.; Gortazar, A.R.; Allen, M.R.; Robling, A.G.; Bouxsein, M.; Schipani, E.; Turner, C.H.; et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 2008, 3, e2942. [Google Scholar] [CrossRef]
- Heiland, G.R.; Zwerina, K.; Baum, W.; Kireva, T.; Distler, J.H.; Grisanti, M.; Asuncion, F.; Li, X.; Ominsky, M.; Richards, W.; et al. Neutralisation of Dkk-1 protects from systemic bone loss during inflammation and reduces sclerostin expression. Ann. Rheum. Dis. 2010, 69, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Unnanuntana, A.; Gladnick, B.P.; Donnelly, E.; Lane, J.M. The assessment of fracture risk. J. Bone Jt. Surg. Am. 2010, 92, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Yuen, N.K.; Ananthakrishnan, S.; Campbell, M.J. Hyperparathyroidism of Renal Disease. Perm. J. 2016, 20, 15–127. [Google Scholar] [CrossRef] [PubMed]
- Andress, D.L. Adynamic bone in patients with chronic kidney disease. Kidney Int. 2008, 73, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Pitt, S.C.; Sippel, R.S.; Chen, H. Secondary and tertiary hyperparathyroidism, state of the art surgical management. Surg. Clin. N. Am. 2009, 89, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Jadoul, M.; Albert, J.M.; Akiba, T.; Akizawa, T.; Arab, L.; Bragg-Gesham, J.L.; Mason, N.; Prutz, K.G.; Young, E.W.; Pisoni, R.L. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2006, 70, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Danese, M.D.; Kim, J.; Doan, Q.V.; Dylan, M.; Griffiths, R.; Chertow, G.M. PTH and the risks for hip, vertebral, and pelvic fractures among patients on dialysis. Am. J. Kidney Dis. 2006, 47, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.; Mawad, H.; El-Husseini, A.A.; Davenport, D.L.; Malluche, H.H. Serum bone markers in ROD patients across the spectrum of decreases in GFR: Activin A increases before all other markers. Clin. Nephrol. 2019, 91, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, H.S.; Behets, G.; Viaene, L.; Bammens, B.; Claes, K.; Meijers, B.; Naesens, M.; Sprangers, B.; Kuypers, D.; Cavalier, E.; et al. Diagnostic Accuracy of Noninvasive Bone Turnover Markers in Renal Osteodystrophy. Am. J. Kidney Dis. 2022, 79, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Graciolli, F.G.; Neves, K.R.; Barreto, F.; Barreto, D.V.; Dos Reis, L.M.; Canziani, M.E.; Sabbagh, Y.; Carvalho, A.B.; Jorgetti, V.; Elias, R.M.; et al. The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017, 91, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Marques, I.D.; Araújo, M.J.; Graciolli, F.G.; Dos Reis, L.M.; Pereira, R.M.; Custodio, R.M.; Jorgetii, V.; Elias, R.M.; David-Neto, E.; Moyses, R.M.A. Biopsy vs. peripheral computed tomography to assess bone disease in CKD patients on dialysis: Differences and similarities. Osteoporos. Int. 2017, 28, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Sprague, S.M.; Bellorin-Font, E.; Jorgetti, V.; Carvalho, A.B.; Malluche, H.H.; Ferreira, A.; D’Haese, P.C.; Drueke, T.B.; Du, H.; Manley, T.; et al. Diagnostic Accuracy of Bone Turnover Markers and Bone Histology in Patients With CKD Treated by Dialysis. Am. J. Kidney Dis. 2016, 67, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Monier-Faugere, M.C.; Magnusson, P.; Malluche, H.H. Bone alkaline phosphatase isoforms in hemodialysis patients with low versus non-low bone turnover: A diagnostic test study. Am. J. Kidney Dis. 2015, 66, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Iimori, S.; Mori, Y.; Akita, W.; Kuyama, T.; Takada, S.; Asai, T.; Kuwahara, M.; Sasaki, S.; Tsukamoto, Y. Diagnostic usefulness of bone mineral density and biochemical markers of bone turnover in predicting fracture in CKD stage 5D patients—A single-center cohort study. Nephrol. Dial. Transplant. 2012, 27, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Julian, B.A.; Laskow, D.A.; Dubovski, J.; Dubovski, E.V.; Curtis, J.J.; Quarles, L.D. Rapid loss of vertebral mineral density after renal transplantation. N. Eng. J. Med. 1991, 325, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Lou, I.; Foley, D.; Odorico, S.K.; Leverson, G.; Schneider, D.F.; Sippel, R.; Chen, H. How Well Does Renal Transplantation Cure Hyperparathyroidism? Ann. Surg. 2015, 262, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Perrin, P.; Caillard, S.; Javier, R.M.; Braun, L.; Borni-Duval, C.; Muller, C.; Olagne, J.; Moulin, B. Persistent hyperparathyroidism is a major risk factor for fractures in the five years after kidney transplantation. Am. J. Transplant. 2013, 13, 2653–2663. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo, G.; Cozzolino, M. FGF23 in kidney transplant: The strange case of Doctor Jekyll and Mister Hyde. Clin. Kidney J. 2016, 9, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: What’s changed and why it matters. Kidney Int. 2017, 92, 26–36. [Google Scholar] [CrossRef]
- Lee, K.; Deeds, J.D.; Segre, G.V. Expression of parathyroid hormone-related peptide and its receptor messenger ribonucleic acids during fetal development of rats. Endocrinology 1995, 136, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Jüppner, H. Molecular cloning and characterization of a parathyroid hormone/parathyroid hormone-related peptide receptor: A member of an ancient family of G protein-coupled receptors. Curr. Opin. Nephrol. Hypertens. 1994, 3, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Colbert, M.C.; Witte, D.; Kuan, C.Y.; Gruenstein, E.; Osinska, H.; Lanske, B.; Kronenberg, H.M.; Clemens, T.L. Midgestational lethality in mice lacking the parathyroid hormone (PTH)/PTH-related peptide receptor is associated with abrupt cardiomyocyte death. Endocrinology 2003, 144, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Crass, F.M. Parathyroid hormone reduces acute ischemic injury of the myocardium. Surg. Gynecol. Obst. 1986, 163, 523–530. [Google Scholar]
- Schlüter, K.; Katzer, C.; Frischkopf, K.; Wenzel, S.; Taimor, G.; Piper, H.M. Expression, release, and biological activity of parathyroid hormone-related peptide from coronary endothelial cells. Circ. Res. 2000, 86, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.K.; Tenner, T.E., Jr.; Yee, J.A.; Yang, M.; Janssen, H.F. Hypotensive action of parathyroid hormone preparations on rats and dogs. Proc. Natl. Acad. Sci. USA 1980, 77, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Lederis, K.; Huang, M.; LeBlanc, F.E.; Rorstad, O.P. Relaxation of bovine, porcine and human brain arteries by parathyroid hormone. Life Sci. 1983, 33, 2497–2503. [Google Scholar] [CrossRef] [PubMed]
- Romero, G.; von Zastrow, M.; Friedman, P.A. Role of PDZ proteins in regulating trafficking, signaling, and function of GPCRs: Means, motif, and opportunity. Adv. Pharmacol. 2011, 62, 279–314. [Google Scholar] [CrossRef]
- Benson, T.; Menezes, T.; Campbell, J.; Bice, A.; Hood, B.; Prisby, R. Mechanisms of vasodilation to PTH 1–84, PTH 1–34, and PTHrP 1–34 in rat bone resistance arteries. Osteoporos. Int. 2016, 27, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Noonan, W.T.; Qian, J.; Stuart, W.D.; Clemens, T.L.; Lorenz, J.N. Altered renal hemodynamics in mice overexpressing the parathyroid hormone (PTH)/PTH-related peptide type 1 receptor in smooth muscle. Endocrinology 2003, 144, 4931–4938. [Google Scholar] [CrossRef]
- Raison, D.; Coquard, C.; Hochane, M.; Steger, J.; Massfelder, T.; Moulin, B.; Karaplis, A.C.; Metzger, D.; Chambon, P.; Helwig, J.J.; et al. Knockdown of parathyroid hormone related protein in smooth muscle cells alters renal hemodynamics but not blood pressure. Am. J. Physiol. Renal Physiol. 2013, 305, F333–F342. [Google Scholar] [CrossRef]
- Brown, S.J.; Ruppe, M.D.; Tabatabai, L.S. The Parathyroid Gland and Heart Disease. Methodist Debakey Cardiovasc. J. 2017, 13, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, M.; Perna, A.F.; Fuiano, G. Secondary Hyperparathyroidism and Hypertension: An Intriguing Couple. J. Clin. Med. 2020, 9, 629. [Google Scholar] [CrossRef]
- Xu, Y.; Evans, M.; Soro, M.; Barany, P.; Carrero, J.J. Secondary hyperparathyroidism and adverse health outcomes in adults with chronic kidney disease. Clin. Kidney J. 2021, 14, 2213–2220. [Google Scholar] [CrossRef]
- Pepe, J.; Cipriani, C.; Sonato, C.; Raimo, O.; Biamonte, F.; Minisola, S. Cardiovascular manifestations of primary hyperparathyroidism: A narrative review. Eur. J. Endocrinol. 2017, 177, R297–R308. [Google Scholar] [CrossRef] [PubMed]
- Nyby, M.D.; Hino, T.; Berger, M.E.; Ormsby, B.L.; Golub, M.S.; Brickman, A.S. Desensitization of vascular tissue to parathyroid hormone and parathyroid hormone-related protein. Endocrinology 1995, 136, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Massfelder, T.; Stewart, A.F.; Endlich, K.; Soifer, N.; Judes, C.; Helwig, J.J. Parathyroid hormone-related protein detection and interaction with NO and cyclic AMP in the renovascular system. Kidney Int. 1996, 50, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Sneddon, W.B.; Syme, C.A.; Bisello, A.; Magyar, C.E.; Rochdi, M.D.; Parent, J.L.; Weinman, E.J.; Abou-Samra, E.J.; Friedman, P.A. Activation-independent parathyroid hormone receptor internalization is regulated by NHERF1 (EBP50). J. Biol. Chem. 2003, 278, 43787–43796. [Google Scholar] [CrossRef] [PubMed]
- Syme, C.A.; Friedman, P.A.; Bisello, A. Parathyroid hormone receptor trafficking contributes to the activation of extracellular signal-regulated kinases but is not required for regulation of cAMP signaling. J. Biol. Chem. 2005, 280, 11281–11288. [Google Scholar] [CrossRef] [PubMed]
- Divieti, P.; Geller, A.I.; Suliman, G.; Jüppner, H.; Bringhurst, F.R. Receptors specific for the carboxyl-terminal region of parathyroid hormone on bone-derived cells: Determinants of ligand binding and bioactivity. Endocrinology 2005, 146, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Hulter, H.N.; Melby, J.C.; Peterson, J.C.; Cooke, C.R. Chronic continuous PTH infusion results in hypertension in normal subjects. J. Clin. Hypertens. 1986, 2, 360–370. [Google Scholar] [PubMed]
- Spät, A.; Hunyady, L. Control of aldosterone secretion: A model for convergence in cellular signaling pathways. Physiol. Rev. 2004, 84, 489–539. [Google Scholar] [CrossRef] [PubMed]
- Isales, C.M.; Barrett, P.Q.; Brines, M.; Bollag, W.; Rasmussen, H. Parathyroid hormone modulates angiotensin II-induced aldosterone secretion from the adrenal glomerulosa cell. Endocrinology 1991, 129, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.; Coureau, C.; Rabin, M.R.; Escoubet, B.; Hruby, M.; Walrant, O.; Silve, C. Parathyroid hormone (PTH)/PTH-related protein receptor messenger ribonucleic acid expression and PTH response in a rat model of secondary hyperparathyroidism associated with vitamin D deficiency. Endocrinology 1995, 136, 3751–3758. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, G.; Aragona, F.; Malendowicz, L.K.; Nussdorfer, G.G. PTH and PTH-related peptide enhance steroid secretion from human adrenocortical cells. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E209–E213. [Google Scholar] [CrossRef] [PubMed]
- Kabadi, U.M. Renal calculi in primary hyperaldosteronism. J. Postgrad. Med. 1995, 41, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Sani, C.; Perazzoli, F.; Casoli, M.C.; Negro, A.; Dotti, C. Alterations of calcium metabolism and of parathyroid function in primary aldosteronism, and their reversal by spironolactone or by surgical removal of aldosterone-producing adenomas. Am. J. Hypertens. 1995, 8, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Pilz, S.; Kienreich, K.; Drechsler, C.; Ritz, E.; Fahrleitner-Pammer, A.; Gaksch, M.; Meinitzer, A.; Marz, W.; Pieber, T.R.; Tomaschitz, A. Hyperparathyroidism in patients with primary aldosteronism: Cross-sectional and interventional data from the GECOH study. J. Clin. Endocrinol. Metab. 2012, 97, E75–E79. [Google Scholar] [CrossRef] [PubMed]
- Atchison, D.K.; Ortiz-Capisano, M.C.; Beierwaltes, W.H. Acute activation of the calcium-sensing receptor inhibits plasma renin activity in vivo. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1020–R1026. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.; Brown, J.M.; Williams, J.S. The renin-angiotensin-aldosterone system and calcium-regulatory hormones. J. Hum. Hypertens. 2015, 29, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.M.; Espiner, E.A.; Nicholls, M.G.; Ikram, H.; Hamilton, E.J.; Maslowski, A.H. Hormone, calcium and blood pressure relationships in primary hyperparathyroidism. J. Hypertens. 1988, 6, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Valvo, E.; Bedogna, V.; Gammaro, L.; Casagrande, P.; Ortalda, V.; Maschio, G. Systemic hemodynamic pattern in primary hyperparathyroidism and its changes after parathyroidectomy. Miner. Electrolyte Metab. 1991, 17, 147–152. [Google Scholar] [PubMed]
- Brown, J.M.; Williams, J.S.; Luther, J.M.; Garg, R.; Garza, A.E.; Pojoga, L.H.; Ruan, D.T.; Williams, G.H.; Adler, G.K.; Vaidya, A. Human interventions to characterize novel relationships between the renin-angiotensin-aldosterone system and parathyroid hormone. Hypertension 2014, 63, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; de Boer, I.H.; Robinson-Cohen, C.; Sicovick, D.S.; Kestenbaum, B.; Allison, M.; Vaidya, A. Aldosterone, parathyroid hormone, and the use of renin-angiotensin-aldosterone system inhibitors: The multi-ethnic study of atherosclerosis. J. Clin. Endocrinol. Metab. 2015, 100, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Yamamoto, H.; Yoshida, K.; Kisanuki, A.; Hirano, Y.; Ohte, N.; Akasaka, T.; Takeuchi, M.; Nakatani, S.; Ohtani, T.; et al. Prognostic factors for progression of early- and late-stage calcific aortic valve disease in Japanese: The Japanese Aortic Stenosis Study (JASS) Retrospective Analysis. Hypertens. Res. 2010, 33, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Yutzey, K.E.; Demer, L.L.; Body, S.C.; Huggins, G.S.; Towler, D.A.; Giachelli, C.M.; Hofmann-Bowman, M.A.; Mortlock, D.P.; Rogers, M.B.; Sadeghi, M.M.; et al. Calcific aortic valve disease: A consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.; Bratt, D.; Lees, J.; DaCosta, M.; Plant, K.; Watson, O.J.; Solaymani-Kohal, S.; Tazzyman, S.; Serbanovic-Canic, J.; Crossman, D.C.; et al. Loss of Function of Parathyroid Hormone Receptor 1 Induces Notch-Dependent Aortic Defects During Zebrafish Vascular Development. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.Y.; Ye, T.; Ling, Q.Y.; Wu, T.; Wu, G.Y.; Zong, G.J. Parathyroid hormone promotes osteoblastic differentiation of endothelial cells via the extracellular signal-regulated protein kinase 1/2 and nuclear factor-κB signaling pathways. Exp. Ther. Med. 2018, 15, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Carrai, P.; Camarri, S.; Pondrelli, C.R.; Gonnelli, S.; Caffarelli, C. Calcification of Cardiac Valves in Metabolic Bone Disease: An Updated Review of Clinical Studies. Clin. Interv. Aging. 2020, 15, 1085–1095. [Google Scholar] [CrossRef]
- Iwata, S.; Hyodo, E.; Yanagi, S.; Hayashi, Y.; Nishiyama, H.; Kamimori, K.; Ota, T.; Matsumura, Y.; Homma, S.; Yoshiyama, M. Parathyroid hormone and systolic blood pressure accelerate the progression of aortic valve stenosis in chronic hemodialysis patients. Int. J. Cardiol. 2013, 163, 256–259. [Google Scholar] [CrossRef]
- Ren, S.C.; Mao, N.; Yi, S.; Ma, X.; Zou, J.Q.; Tang, X.; Fan, J.M. Vascular Calcification in Chronic Kidney Disease: An Update and Perspective. Aging Dis. 2022, 13, 673–697. [Google Scholar] [CrossRef]
- Block, G.A.; Bushinsky, D.A.; Cheng, S.; Cunningham, J.; Dehmel, B.; Drueke, T.B.; Kettler, M.; Kewalramani, R.; Martin, K.J.; Moe, S.M.; et al. Effect of Etelcalcetide vs Cinacalcet on Serum Parathyroid Hormone in Patients Receiving Hemodialysis With Secondary Hyperparathyroidism: A Randomized Clinical Trial. JAMA 2017, 317, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Tang, R.N.; Liu, H.; Pan, M.M.; Liu, B.C. Cinacalcet ameliorates aortic calcification in uremic rats via suppression of endothelial-to-mesenchymal transition. Acta Pharmacol. Sin. 2016, 37, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Tokunaga, S.; Kawai, M.; Murai, M.; Kobayashi, M.; Kitayama, T.; Saeki, S.; Kawata, T. Evocalcet prevents ectopic calcification and parathyroid hyperplasia in rats with secondary hyperparathyroidism. PLoS ONE 2020, 15, e0232428. [Google Scholar] [CrossRef] [PubMed]
- Unger, P.; Clavel, M.A.; Lindman, B.R.; Mathieu, P.; Pibarot, P. Pathophysiology and management of multivalvular disease. Nat. Rev. Cardiol. 2016, 13, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Bilezikian, J.P. Primary Hyperparathyroidism. J. Clin. Endocrinol. Metab. 2018, 103, 3993–4004. [Google Scholar] [CrossRef] [PubMed]
- Prisby, R.; Menezes, T.; Campbell, J. Vasodilation to PTH (1–84) in bone arteries is dependent upon the vascular endothelium and is mediated partially via VEGF signaling. Bone 2013, 54, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Prisby, R.; Guignandon, A.; Vanden-Bossche, A.; Mac-Way, F.; Linossier, M.T.; Thomas, M.; Laroche, N.; Malaval, L.; Langer, M.; Peter, Z.A.; et al. Intermittent PTH(1–84) Is Osteoanabolic but Not Osteoangiogenic and Relocates Bone Marrow Blood Vessels Closer to Bone-Forming Sites. J. Bone Miner. Res. 2011, 26, 2579–2582. [Google Scholar] [CrossRef] [PubMed]
- Rashid, G.; Bernheim, J.; Green, J.; Benchetrit, S. Parathyroid hormone stimulates the endothelial expression of vascular endothelial growth factor. Eur. J. Clin. Investig. 2008, 38, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Gohin, S.; Carriero, A.; Chenu, C.; Pitsillides, A.A.; Arnett, T.R.; Marenzana, M. The anabolic action of intermittent parathyroid hormone on cortical bone depends partly on its ability to induce nitric oxide-mediated vasorelaxation in BALB/c mice. Cell Biochem. Funct. 2016, 34, 52–62. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Marchais, S.J.; Guérin, A.P.; de Vernejoul, M.C. Ankle-brachial index and bone turnover in patients on dialysis. J. Am. Soc. Nephrol. 2015, 26, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.C.; Ewing, S.K.; Diem, S.J.; Taylor, B.C.; Orwoll, E.S.; Cummings, R.S.; Strotmeyer, E.S.; Ensrud, K.E.; Osteoporotic Fractures in Men (MrOS) Study Group. Peripheral arterial disease is associated with higher rates of hip bone loss and increased fracture risk in older men. Circulation 2009, 119, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Saliba, W.; El-Haddad, B. Secondary hyperparathyroidism: Pathophysiology and treatment. J. Am. Board Fam. Med. 2009, 22, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Frazão, J.M.; Monier-Faugere, M.C.; Gil, C.; Galvao, J.; Oliveira, C.; Baldaia, J.; Rodrigues, I.; Santos, C.; Ribeiro, S.; et al. Effects of sevelamer hydrochloride and calcium carbonate on renal osteodystrophy in hemodialysis patients. J. Am. Soc. Nephrol. 2008, 19, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Slatopolsky, E.A.; Burke, S.K.; Dillon, M.A. RenaGel, a nonabsorbed calcium- and aluminum-free phosphate binder, lowers serum phosphorus and parathyroid hormone. The RenaGel Study Group. Kidney Int. 1999, 55, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Gutiérrez, O.M.; Chang, Y.; Shah, A.; Tamez, H.; Smith, K.; Thadhani, R.; Wolf, M. Phosphorus binders and survival on hemodialysis. J. Am. Soc. Nephrol. 2009, 20, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Gallant, H.K.M.; Spiegel, D.M. Calcium Balance in Chronic Kidney Disease. Curr. Osteoporos. Rep. 2017, 15, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Levi, R.; Ben-Dov, I.Z.; Lavi-Moshayoff, V.; Dinur, M.; Martin, D.; Naveh-Many, T.; Silver, J. Increased parathyroid hormone gene expression in secondary hyperparathyroidism of experimental uremia is reversed by calcimimetics: Correlation with posttranslational modification of the trans acting factor AUF1. J. Am. Soc. Nephrol. 2006, 17, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Eastell, R.; Brandi, M.L.; Costa, A.G.; D’Amour, P.; Shoback, D.M.; Thakker, R.V. Diagnosis of asymptomatic primary hyperparathyroidism: Proceedings of the Fourth International Workshop. J. Clin. Endocrinol. Metab. 2014, 99, 3570–3579. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, Y.; Okano, K.; Kikuchi, K.; Tsuruta, Y.; Akiba, T.; Nitta, K. Effects of cinacalcet on bone mineral density and bone markers in hemodialysis patients with secondary hyperparathyroidism. Clin. Exp. Nephrol. 2013, 17, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Chertow, G.M.; Parfrey, P.S.; Kubo, Y.; Block, G.A.; Correa-Rotter, R.; Drueke, T.B.; Herzog, C.A.; London, G.M.; Mahaffey, K.W.; et al. Cinacalcet, Fibroblast Growth Factor-23, and Cardiovascular Disease in Hemodialysis The Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) Trial. Circulation 2015, 132, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Cruzado, J.M.; Moreno, P.; Torregrosa, J.V.; Taco, O.; Mast, R.; Gomez-Vaquero, C.; Polo, C.; Revuelta, I.; Francos, J.; Torras, J.; et al. A Randomized Study Comparing Parathyroidectomy with Cinacalcet for Treating Hypercalcemia in Kidney Allograft Recipients with Hyperparathyroidism. J. Am. Soc. Nephrol. 2016, 27, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Haris, A.; Sherrard, D.J.; Hercz, G. Reversal of adynamic bone disease by lowering of dialysate calcium. Kidney Int. 2006, 70, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Lips, P.; Goldsmith, D.; de Jongh, R. Vitamin D and osteoporosis in chronic kidney disease. J. Nephrol. 2017, 30, 671–675. [Google Scholar] [CrossRef]
- Jean, G.; Souberbielle, J.C.; Chazot, C. Vitamin D in Chronic Kidney Disease and Dialysis Patients. Nutrients 2017, 9, 328. [Google Scholar] [CrossRef] [PubMed]
- Coyne, D.W.; Goldberg, S.; Faber, M.; Ghossein, C.; Sprague, S.M. A randomized multicenter trial of paricalcitol versus calcitriol for secondary hyperparathyroidism in stages 3–4 CKD. Clin. J. Am. Soc. Nephrol. 2014, 9, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.A.U.; Cohen-Solal, M. Evaluation of fracture risk in chronic kidney disease. J. Nephrol. 2017, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Wang, J.; Song, M.; Giovannucci, E.L.; Ma, H.; Jin, G.; Hu, Z.; Shen, H.; Hang, D. Vitamin D Status and Risk of All-Cause and Cause-Specific Mortality in a Large Cohort: Results From the UK Biobank. J. Clin. Endocrinol. Metabol. 2020, 105, 10. [Google Scholar] [CrossRef] [PubMed]
- Yeung, W.C.G.; Palmer, S.C.; Strippoli, G.F.M.; Talbot, B.; Shah, N.; Hawley, C.M.; Toussaint, N.D.; Badve, S.V. Vitamin D Therapy in Adults With CKD: A Systematic Review and Meta-analysis. Am. J. Kidney Dis. 2023, 82, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Sumida, K.; Ubara, Y.; Hoshino, J.; Mise, K.; Hayami, N.; Suwabe, T.; Kawada, M.; Imafuku, A.; Hiramatsu, R.; Hasewaga, E.; et al. Once-weekly teriparatide in hemodialysis patients with hypoparathyroidism and low bone mass: A prospective study. Osteoporos. Int. 2016, 27, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.D.; Schwartz, E.N.; Chen, P.; Misurski, D.A.; Krege, J.H. Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporos. Int. 2006, 18, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, A.; Yoshiki, F.; Taketsuna, M.; Kajimoto, K.; Enomoto, H. Safety and effectiveness of daily teriparatide for osteoporosis in patients with severe stages of chronic kidney disease: Post hoc analysis of a postmarketing observational study. Clin. Interv. Aging. 2016, 11, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Lo, W.C.; Hu, P.J.; Chan, H.C.; Shen, W.C.; Wu, M.S.; Wu, M.Y. Efficacy of Osteoporosis Medications for Patients with Chronic Kidney Disease: An Updated Systematic Review and Network Meta-Analysis. Front. Pharmacol. 2022, 13, 822178. [Google Scholar] [CrossRef] [PubMed]
- Pazianas, M.; Miller, P.D. Osteoporosis and Chronic Kidney Disease–Mineral and Bone Disorder (CKD-MBD): Back to Basics. Am. J. Kidney Dis. 2021, 78, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Diao, Z.; Wang, Y.; Zhou, P.; Ding, R.; Liu, W. Hemodialysis patients with low serum parathyroid hormone levels have a poorer prognosis than those with secondary hyperparathyroidism. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820958322. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Brancaccio, D.; Cannella, G.; Messa, P.; Gesualdo, L.; Marangella, M.; LoDeserto, C.; Pozzato, M.; Rombola, G.; Costanzo, A.M.; et al. VDRA therapy is associated with improved survival in dialysis patients with serum intact PTH ≤ 150 pg/mL: Results of the Italian FARO Survey. Nephrol. Dial. Transplant. 2012, 27, 3588–3594. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Ciceri, P. Serum PTH levels in dialysis: Better safe than sorry. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820974172. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Wu, S.; Juppner, H.; Green, J.; Aragay, A.M.; Fagin, J.A.; Clemens, T.L. Cell-Specific Signal Transduction of Parathroid Hormone (PTH)-Related Protein through Stably Expressed Recombinant PTH/PTHrP Receptors in Vascular Smooth Muscle Cells. Endocrinology 1996, 137, 3154–3162. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, K.D.; Weber, M.; Piper, H.M. Parathyroid hormone induces protein kinase C but not adenylate cyclase in adult cardiomyocytes and regulates cyclic AMP levels via protein kinase C-dependent phosphodiesterase activity. Biochem. J. 1995, 310, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Friedman, P.A.; Coutermarsh, B.A.; Kennedy, S.M.; Gesek, F.A. Parathyroid hormone stimulation of calcium transport is mediated by dual signaling mechanisms involving protein kinase A and protein kinase C. Endocrinology 1996, 137, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, Y.; Friedman, P.A. Na/H exchange regulatory factor 1, a novel AKT-associating protein, regulates extracellular signal-regulated kinase signaling through a B-Raf-mediated pathway. Mol. Biol. Cell 2008, 19, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Mahon, M.J.; Donowitz, M.; Yun, C.C.; Segre, G.V. Na+/H+ exchanger regulatory factor 2 directs parathyroid hormone 1 receptor signalling. Nature 2002, 417, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, Y.; Abou-Samra, A.B.; Friedman, P.A. NHERF1 Regulates Parathyroid Hormone Receptor Desensitization: Interference with -Arrestin Binding. Mol. Pharmacol. 2009, 75, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Karim, Z.; Gérard, B.; Bakouh, N.; Alili, R.; Leroy, C.; Beck, L.; Silve, C.; Planelles, G.; Urena-Torres, P.; Grandchamp, B.; et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N. Engl. J. Med. 2008, 359, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Ren, H.; Wang, H.; Jose, P.A.; Zeng, C.; Xia, T.; Yang, J. Effect of D4 Dopamine Receptor on Na+-K+-ATPase Activity in Renal Proximal Tubule Cells. Cardiol. Discov. 2023, 3, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Barati, M.T.; Ketchem, C.J.; Merchant, M.L.; Kusiak, W.B.; Jose, P.A.; Weinman, E.J.; LeBlanc, A.J.; Lederer, E.D.; Khundmiri, S.J. Loss of NHERF-1 expression prevents dopamine-mediated Na-K-ATPase regulation in renal proximal tubule cells from rat models of hypertension: Aged F344 rats and spontaneously hypertensive rats. Am. J. Physiol. Cell Physiol. 2017, 313, C197–C206. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Gu, Y.; Zhao, Z.; Huang, J.; Jiang, W.G.; Cheng, S. NHERF1 Suppresses Lung Cancer Cell Migration by Regulation of Epithelial-Mesenchymal Transition. Anticancer Res. 2017, 37, 4405–4414. [Google Scholar] [CrossRef] [PubMed]
- Bushau-Sprinkle, A.; Barati, M.; Conklin, C.; Dupre, T.; Gagnon, K.B.; Khundmiri, S.J.; Clark, B.; Siskind, L.; Doll, M.A.; Rane, M.; et al. Loss of the Na+/H+ Exchange Regulatory Factor 1 Increases Susceptibility to Cisplatin-Induced Acute Kidney Injury. Am. J. Pathol. 2019, 189, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Pushpakumar, S.; Ketchem, C.J.; Sen, U.; Lederer, E.D.; Khundmiri, S.J. Role of Sodium Hydrogen Exchange Regulatory Factor-1 (NHERF1) in Aging: Tale of Two Rat Models. Physiology 2018, 31, 857. [Google Scholar] [CrossRef]
- Gagnon, K.B.; Bushau-Sprinkle, A.; Hu, M.C.; Chang, A.N.; Miller, R.T.; Eleanor, L.D. Kidney Injury Stimulates Apical Translocation of NHERF1. Physiology 2022, 36. [Google Scholar] [CrossRef]
- Bushau-Sprinkle, A.M.; Lederer, E.D. New roles of the Na+/H+ exchange regulatory factor 1 scaffolding protein: A review. Am. J. Physiol. Renal Physiol. 2020, 318, F804–F808. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maranduca, M.A.; Cozma, C.T.; Clim, A.; Pinzariu, A.C.; Tudorancea, I.; Popa, I.P.; Lazar, C.I.; Moscalu, R.; Filip, N.; Moscalu, M.; et al. The Molecular Mechanisms Underlying the Systemic Effects Mediated by Parathormone in the Context of Chronic Kidney Disease. Curr. Issues Mol. Biol. 2024, 46, 3877-3905. https://doi.org/10.3390/cimb46050241
Maranduca MA, Cozma CT, Clim A, Pinzariu AC, Tudorancea I, Popa IP, Lazar CI, Moscalu R, Filip N, Moscalu M, et al. The Molecular Mechanisms Underlying the Systemic Effects Mediated by Parathormone in the Context of Chronic Kidney Disease. Current Issues in Molecular Biology. 2024; 46(5):3877-3905. https://doi.org/10.3390/cimb46050241
Chicago/Turabian StyleMaranduca, Minela Aida, Cristian Tudor Cozma, Andreea Clim, Alin Constantin Pinzariu, Ionut Tudorancea, Irene Paula Popa, Cristina Iuliana Lazar, Roxana Moscalu, Nina Filip, Mihaela Moscalu, and et al. 2024. "The Molecular Mechanisms Underlying the Systemic Effects Mediated by Parathormone in the Context of Chronic Kidney Disease" Current Issues in Molecular Biology 46, no. 5: 3877-3905. https://doi.org/10.3390/cimb46050241
APA StyleMaranduca, M. A., Cozma, C. T., Clim, A., Pinzariu, A. C., Tudorancea, I., Popa, I. P., Lazar, C. I., Moscalu, R., Filip, N., Moscalu, M., Constantin, M., Scripcariu, D. V., Serban, D. N., & Serban, I. L. (2024). The Molecular Mechanisms Underlying the Systemic Effects Mediated by Parathormone in the Context of Chronic Kidney Disease. Current Issues in Molecular Biology, 46(5), 3877-3905. https://doi.org/10.3390/cimb46050241