Congenital Hypopigmentary Disorders with Multiorgan Impairment: A Case Report and an Overview on Gray Hair Syndromes




Abstract
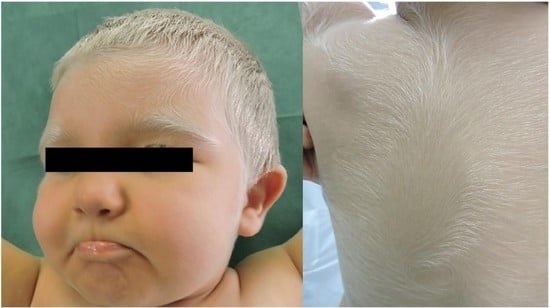
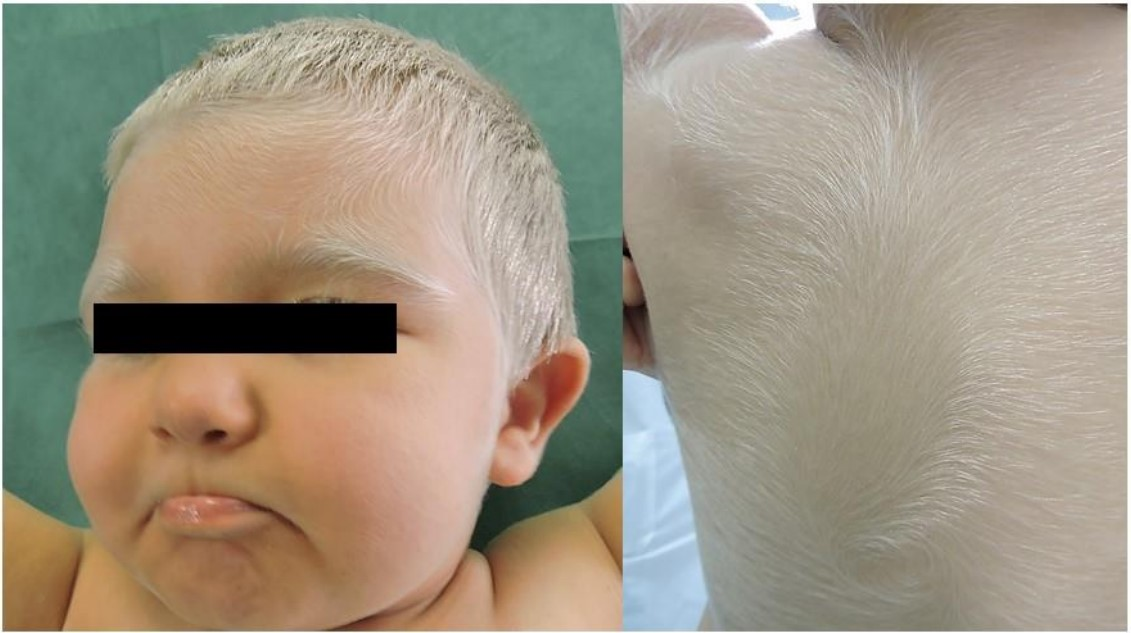
:
1. Introduction
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tey, H.L. A practical classification of childhood hypopigmentation disorders. Acta Derm. Venereol. 2010, 90, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Sahu, C.; Netam, S.S.; Bhutada, B.R.; Jaiswal, S.J. Griscelli syndrome: A rare disorder. Neurol. India 2017, 65, 869–870. [Google Scholar] [PubMed]
- Minocha, P.; Choudhary, R.; Agrawal, A.; Sitaraman, S. Griscelli syndrome subtype 2 with hemophagocytic lympho-histiocytosis: A case report and review of literature. Intractable Rare Dis. Res. 2017, 6, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.J.; Jagati, A.K.; Katrodiya, N.K.; Patel, S.M. Griscelli syndrome type-3. Indian Dermatol. Online J. 2016, 7, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Chandravathi, P.L.; Karani, H.D.; Siddaiahgari, S.R.; Lingappa, L. Light Microscopy and Polarized Microscopy: A Dermatological Tool to Diagnose Gray Hair Syndromes. Int. J. Trichol. 2017, 9, 38–41. [Google Scholar]
- Mohammadzadeh Shanehsaz, S.; Rezazadeh, A.; Dandashli, A. Elejalde syndrome (ES). Dermatol. Online J. 2015, 2, 13. [Google Scholar]
- De Almeida, H.L., Jr.; Kiszewski, A.E.; Vicentini Xavier, T.; Pirolli, F.; Antônio Suita de Castro, L.A. Ultrastructural aspects of hairs of Chediak-Higashi syndrome. J. Eur. Acad. Dermatol. Venereol. 2018, 32, e227–e229. [Google Scholar] [CrossRef] [PubMed]
- Grandin, V.; Sepulveda, F.E.; Lambert, N.; Al Zahrani, M.; Al Idrissi, E.; Al-Mousa, H.; Almanjomi, F.; Al-Ghonaium, A.; Habazi, M.; Alghamdi, H.; et al. RAB27A duplication in several cases of Griscelli syndrome type 2: An explanation for cases lacking a genetic diagnosis. Hum. Mutat. 2017, 38, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Prevalence of Rare Diseases: Bibliographic Data, Prevalence, Incidence or Number of Published Cases Listed by Diseases (In Alphabetical Order). Available online: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_diseases.pdf (accessed on 24 February 2019).
- Bizario, J.C.; Feldmann, J.; Castro, F.A.; Ménasché, G.; Jacob, C.M.; Cristofani, L.; Casella, E.B.; Voltarelli, J.C.; de Saint-Basile, G.; Espreafico, E.M. Griscelli syndrome: Characterization of a new mutation and rescue of T-cytotoxic activity by retroviral transfer of RAB27A gene. J. Clin. Immunol. 2004, 24, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Chabchoub, E.; Cogulu, O.; Durmaz, B.; Vermeesch, J.R.; Ozkinay, F.; Fryns, J.P. Oculocerebral hypopigmentation syndrome maps to chromosome 3q27.1q29. Dermatology 2011, 223, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.P.; Booker, R.C.; Morrison, S.J.; Le, L.Q. Identification of hair shaft progenitors that create a niche for hair pigmentation. Genes Dev. 2017, 31, 744–756. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Syndrome | Clinical Findings | Genetic Findings | Number of Cases Reported Until Now/Epidemiology [11] | |
|---|---|---|---|---|
| Dermatological Signs | Nondermatological Signs | |||
| GS Griscelli syndrome GS, type 1 (GS1) GS, type 2 (GS2) GS, type 3 (GS3) | GS Hair: Silvery scalp hair, eyebrows, and eyelashes GS Skin: Generalized hypopigmentation with tanning capacities after sun exposure | GS1 Neurologic defects epilepsy, endocranic hypertension, cerebellar signs, hemiparesis, spasticity, hypotonia, peripheral facial palsy, psychomotor retardation Eyes Blue to brown iris | GS1 MYO5A (15q21.2) | GS1 20 cases |
| GS2 Immunological defects absence of delayed-type cutaneous hypersensitivity, impaired NK cell function, hypogammaglobulinemia, hemofagocitic lymphohystiocitosis, accelerated phases of hematological disease Neurological defects progressive neurological deterioration due to cerebral macrophage infiltration Eyes Blue to brown iris | GS2 RAB27A (15q21.3) | GS2 102 cases | ||
| GS3 no other systemic signs | GS3 MLPH (2q37.3) | GS3 13 cases | ||
| CHS Chediak–Higashi syndrome | CHS Hair: Silvery scalp hair, eyebrows, and eyelashes CHS Skin: Fair skin with tanning capacities (possible hyperpigmentation after sun exposure) | CHS Ocular signs possible strabismus, nystagmus, photophobia Immunological dysfunction recurrent infections (impaired chemotaxis of granulocytes, diminished antibody-dependent cytotoxicity, defective NK cell function); uncontrolled macrophage activation and lymphohistiocytic infiltrates into major organs (about 85% develop HLH) Neurological defects progressive neurological deterioration (cerebral macrophage infiltration) Eyes Blue to brown iris | CHS AR LYST (1q42-43) | 600 cases |
| ES Elejalde syndrome (neuroectodermal melanolysosomal disease) | ES Hair: Leaden-to-silvery scalp hair, eyebrows, and eyelashes ES Skin: Generalized hypopigmentation with intense tanning (bronze skin color) after sun exposure | ES Neurologic defects Like GS1 Ocular signs nystagmus, diplopia, congenital amaurosis Other findings omphalocele | ES AR MYO5A (15q21.2) | 20 cases |
| OHS Oculocerebral hypopigmentation syndrome, Cross type | OHS Hair: Silvery scalp hair, eyebrows, and eyelashes OHS Skin: Generalized hypopigmentation (very light) with photosensitivity | OHS Neurological signs growth deficiency, intellectual disability, spastic tetraplegia, athetoid movements, hyperreflexia Ocular signs microphthalmia, corneal and lens opacity, ectropium, nystagmus, optic nerve atrophy, visual impairment/blindness Other findings Abnormal palate morphology, gingival fibromatosis, dolichocephaly, oligophrenia | OHS AR gene unknown (3q27.1q29) | 14 cases |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gironi, L.C.; Zottarelli, F.; Savoldi, G.; Notarangelo, L.D.; Basso, M.E.; Ferrero, I.; Timeus, F.; Fagioli, F.; Maiuri, L.; Colombo, E.; et al. Congenital Hypopigmentary Disorders with Multiorgan Impairment: A Case Report and an Overview on Gray Hair Syndromes. Medicina 2019, 55, 78. https://doi.org/10.3390/medicina55030078
Gironi LC, Zottarelli F, Savoldi G, Notarangelo LD, Basso ME, Ferrero I, Timeus F, Fagioli F, Maiuri L, Colombo E, et al. Congenital Hypopigmentary Disorders with Multiorgan Impairment: A Case Report and an Overview on Gray Hair Syndromes. Medicina. 2019; 55(3):78. https://doi.org/10.3390/medicina55030078
Chicago/Turabian StyleGironi, Laura Cristina, Francesca Zottarelli, Gianfranco Savoldi, Lucia Dora Notarangelo, Maria Eleonora Basso, Ivana Ferrero, Fabio Timeus, Franca Fagioli, Luigi Maiuri, Enrico Colombo, and et al. 2019. "Congenital Hypopigmentary Disorders with Multiorgan Impairment: A Case Report and an Overview on Gray Hair Syndromes" Medicina 55, no. 3: 78. https://doi.org/10.3390/medicina55030078
APA StyleGironi, L. C., Zottarelli, F., Savoldi, G., Notarangelo, L. D., Basso, M. E., Ferrero, I., Timeus, F., Fagioli, F., Maiuri, L., Colombo, E., & Savoia, P. (2019). Congenital Hypopigmentary Disorders with Multiorgan Impairment: A Case Report and an Overview on Gray Hair Syndromes. Medicina, 55(3), 78. https://doi.org/10.3390/medicina55030078