2. Case Report and Results
A 50-year-old woman (written consent was obtained from the patient and the faculty ethics committee (20-047KB)) presented in our clinic in December 2015 with diffuse bone, muscle, and joint pain. She reported the first symptoms about a year earlier and an aggravation ever since. During that time, she had consulted several outpatient physicians. Dual energy X-ray absorptiometry had shown osteopenia with a minimal T-score of −2.0 standard deviations at the lumbar spine. A skeletal scintigraphy had displayed nuclear activity enrichments in the right ribs, thoracic spine, hips, and facial bones. Menopause started in 2014 and the patient received hormone replacement therapy for six months before the presentation in our clinic. Further therapy included cholecalciferol and intravenous bisphosphonates, the latter under the assumption of a transitory osteoporosis of the left hip. Secondary causes of osteoporosis (i.e., hypercortisolism, mastocytosis, monoclonal gammopathy, rheumatological disorder, gynaecological tumor or other malignancy, Paget’s disease) had been excluded. Since no plausible explanation had been found for the findings and symptoms, the complaints had been interpreted as a consequence of degenerative spinal changes.
The patient eventually presented in our clinic with new pain in the right knee and left foot. MRI imaging revealed a fracture of the first left metatarsal bone with concomitant oedema, oedema without fracture in the spongiosa of the medial talus and the os cuneiforme laterale, as well as infractions in the right inferior patella epiphysis and the anteromedial tibia. In the short run, the patient developed progressive pain in both knees and became unable to walk. The MRI scan now displayed ostechondrosis dissecans of the left medial talus plus various fractures (medial metaphysis of the right tibia head, medial femur condyles, distal cuboid and proximal os cuneiforme). The patient denied an adequate trauma or maltreatment.
Laboratory analysis revealed normocalcemia, but considerable hypophosphatemia (1.6 mg/dL; range: 2.5–4.8 mg/dL) and a moderately elevated alkaline phosphatase (114 U/L; range: 35–105 U/L) and parathyroid hormone (185 pg/mL; range: 15–65 pg/mL). Retrospective resurvey of external laboratory values also showed decreased phosphate levels, albeit not consistently that distinct. The 25-OH-vitamin D3 level was normal (39 ng/mL; range 20–60 ng/mL), whereas 1,25-OH-vitamin D3 was decreased (13 ng/L; range 25–86 ng/L). Urinary analysis displayed a low phosphate value (17.9 mg/dL; reference 22–74 mg/dL). However, fractional phosphate excretion (48%; reference 5–20%) and phosphate clearance (44 mL/min; reference 5.4–16.2 mL/min) were increased. C-terminal FGF-23 was elevated (224 kRU/L; reference 26–111 kRU/L).
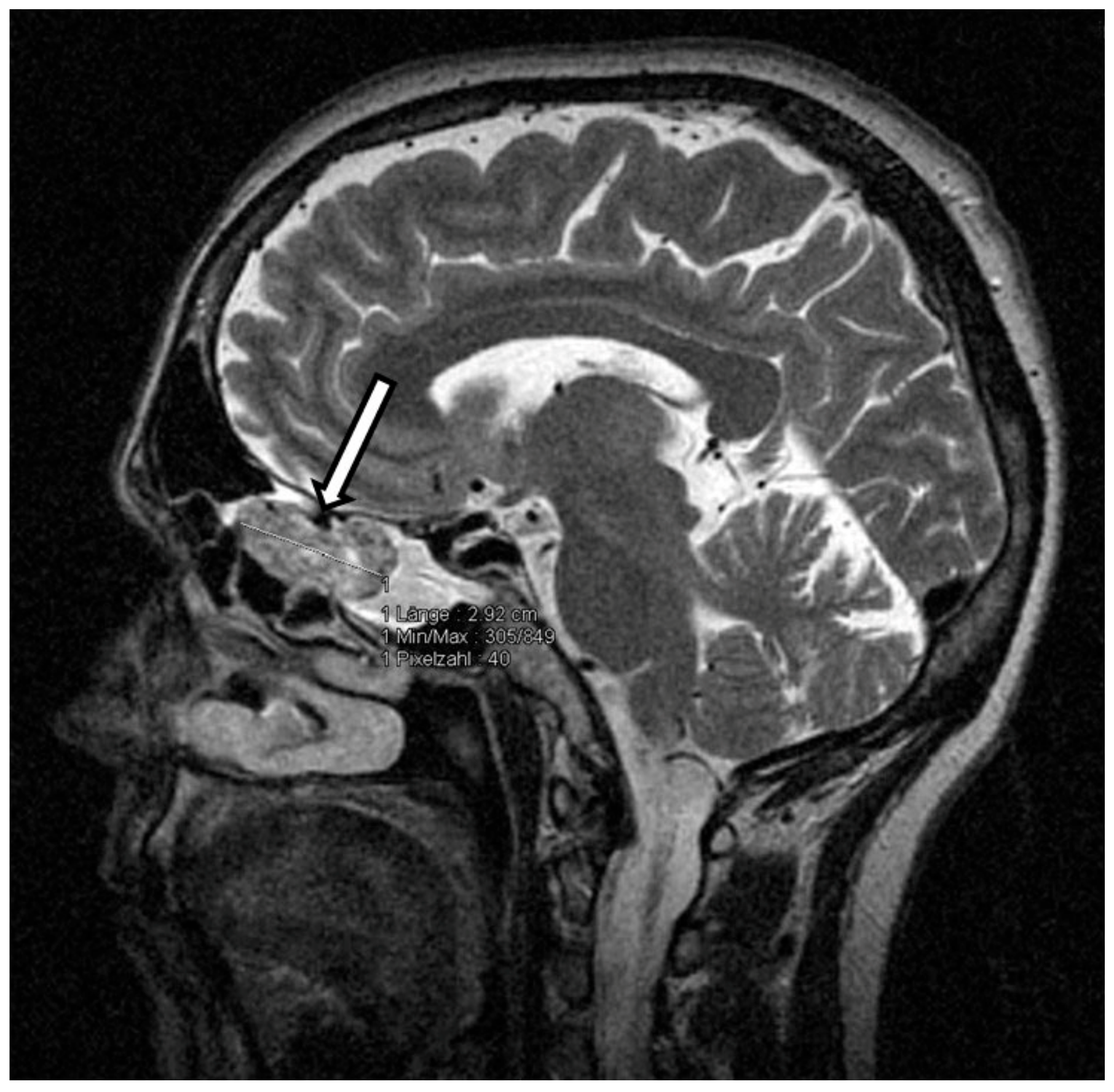
Thus, oncogenic osteomalacia due to neoplastic FGF-23 secretion was suspected. FDG-PET-CT and subsequently DOTATATE-PET-CT imaging demonstrated no tumor. Since the cranium was not represented in both PET-CT scans, contrast enhanced cranial MRI was performed and finally revealed a tumorous mass in the left cellulae ethmoidales with an expansion of 3.2 cm × 1.4 cm × 1.8 cm and intensive contrast enhancement (
Figure 1). Remarkably, retrospective analysis of an MRI scan of the head, which had been performed months earlier in an outpatient clinic, already showed the tumor, which had not been described back then. Even in retrospect, our patient denied symptoms possibly related to a tumor located in the cellulae ethmoidales, such as nasal obstruction, epistaxis, hyposmia, diplopia, tuba eustachii obstruction, or frontal headache.
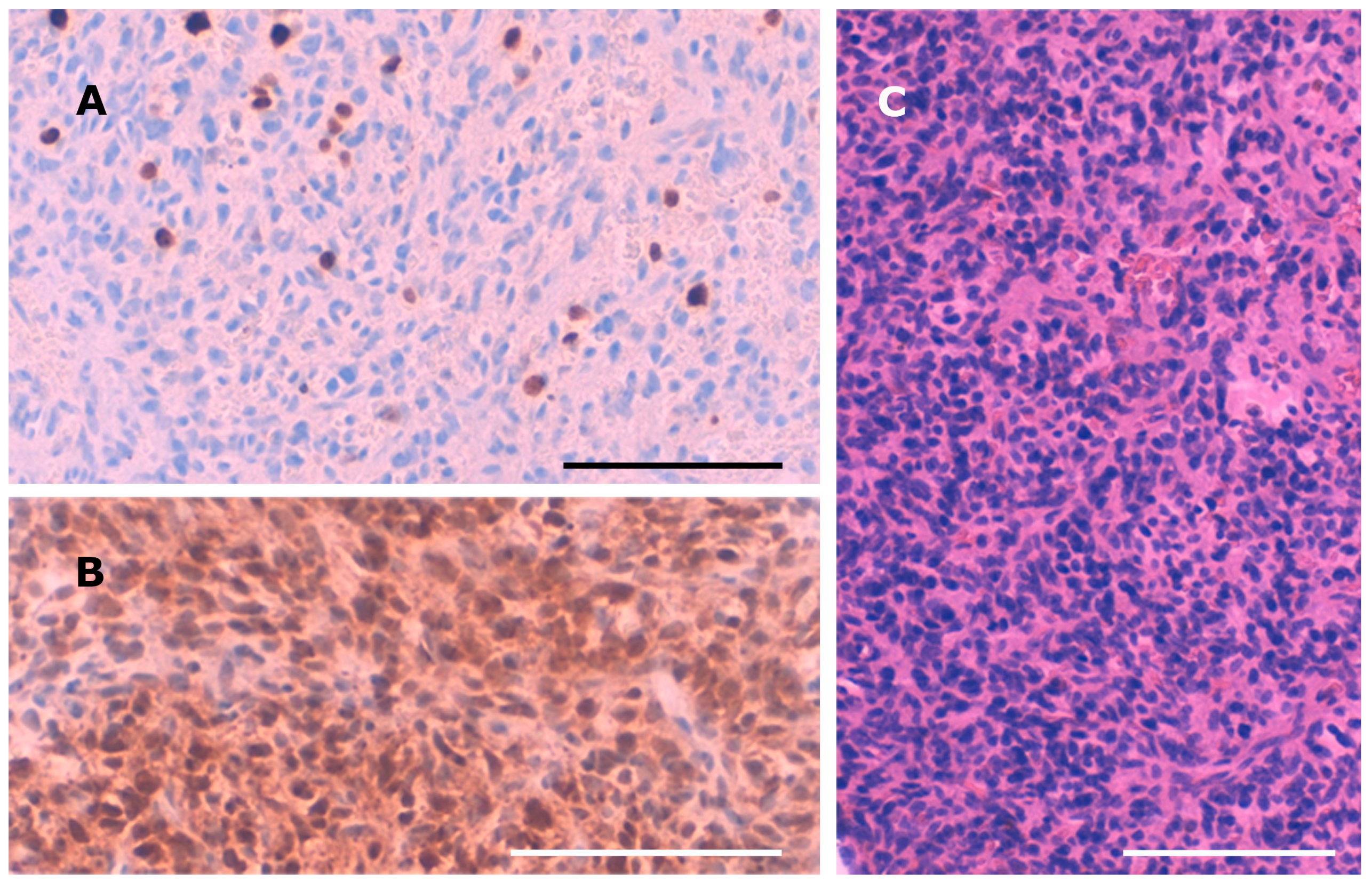
Intraoperatively, the tumor presented as a bleeding, amorphous mass with bony erosion of the posterior ethmoidal cells. It was fully resected in piecemeal technique via expanded endonasal surgery including the left cribriform plate as well as the left papyraceous membrane. The skull base defect was successfully closed using a pedicled nasoseptal flap. Histopathological examination showed a neuron-specific-enolase-positive, cell-rich but poorly proliferating (Ki67: 5%) tumor (
Figure 2). The nuclei were moderately pleomorphic. Pseudorosettes were found in some tumor areas. No necrotic areas were observed. Staining for CD3, CD20, CD45, GFAP, CD31, S100, STAT6, actin, HMB45, CD99, EMA, TLE, synaptophysin, chromogranin, CD34, CD68, KL1, desmin, and ceratin was negative. Only scarce tumor matrix was observed, while hyalinized matrix would be a leading feature for the diagnosis of a phosphaturic mesenchymal tumor (PMT). The tumor showed a well-developed capillary network, though perocytoma-like pattern or haemangioma-like pattern was not observed. Calcification was not seen. The location in the cellulae ethmoidales and the histopathological features of the tumor first led to the diagnosis of an olfactory neuroblastoma. However, additional staining for SATB2 and beta-Catenin revealed nuclear positivity for SATB2 and nuclear negativity for beta-Catenin, confirming the diagnosis of PMT.
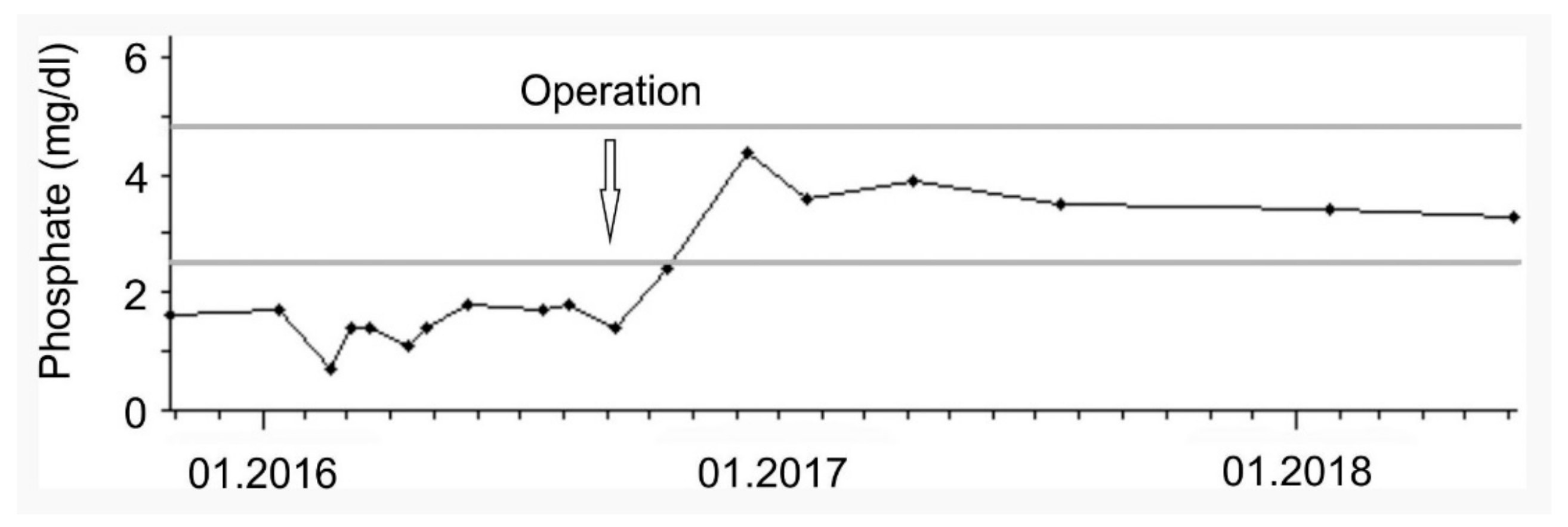
Following the resection, FGF-23 (63 kRU/L), parathyroid hormone, and phosphate levels (
Figure 3) normalized and phosphate supplementation was terminated. The patient described a significant improvement of her painful symptoms and general condition and regained the ability to walk and participate in daily life activities without restrictions.
3. Discussion
PMTs are rare mesenchymal tumors that are claimed to be morphologically distinctive neoplasms [
3]. Due to the rarity of the disease, the prevalence of oncogenic osteomalacia is not known. The literature and Orphanet report approximately 400 cases worldwide to date [
1,
4]. Most tumors occur in middle-aged adults. Often, patients are diagnosed late, with a long history of osteomalacia, which was also true for our patient. The reported case demonstrates several difficulties that can occur when dealing with an orphan disease. Hypophosphatemia had been documented months before the final diagnosis was established. Since calcium levels had been normal and parathyroid hormone levels were only inconsistently elevated, too little attention was paid to this single laboratory parameter, which is often only attended in context with changes in calcium levels or when being elevated in chronic kidney disease. Further, once suspected, localizing a PMT may be challenging. PMTs are typically benign, slowly growing, and therefore often small and without local symptoms. Furthermore, they do not have a predilection site, but may occur anywhere in the body, including unexpected locations, such as hands and soles of the feet [
5]. In our case, the tumor itself had been pictured months before the final diagnosis, but was not identified as such, probably because the MRI scan was performed in order to detect skeletal changes and a soft tissue tumor was not expected. Further, although we were aware of the necessity of a whole-body scan when searching for an FGF-23-producing tumor and also communicated this to our radiology department, the cranium was not pictured in both performed PET-CT scans. Finally, the unusual location and morphology of the tumor led to the initial diagnosis of an olfactory neuroblastoma that was later revised to PMT after additional immunohistochemical analyses.

 ,
, 
{kind=link}
{kind=link}
{kind=link}


