Systemic Oxidative Stress Markers in Cirrhotic Patients with Hepatic Encephalopathy: Possible Connections with Systemic Ammoniemia

 ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Material and Methods
2.1. Patients and Healthy Subjects
2.2. Measurement of Oxidative Stress Markers
2.3. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Oxidative Stress-Related Markers
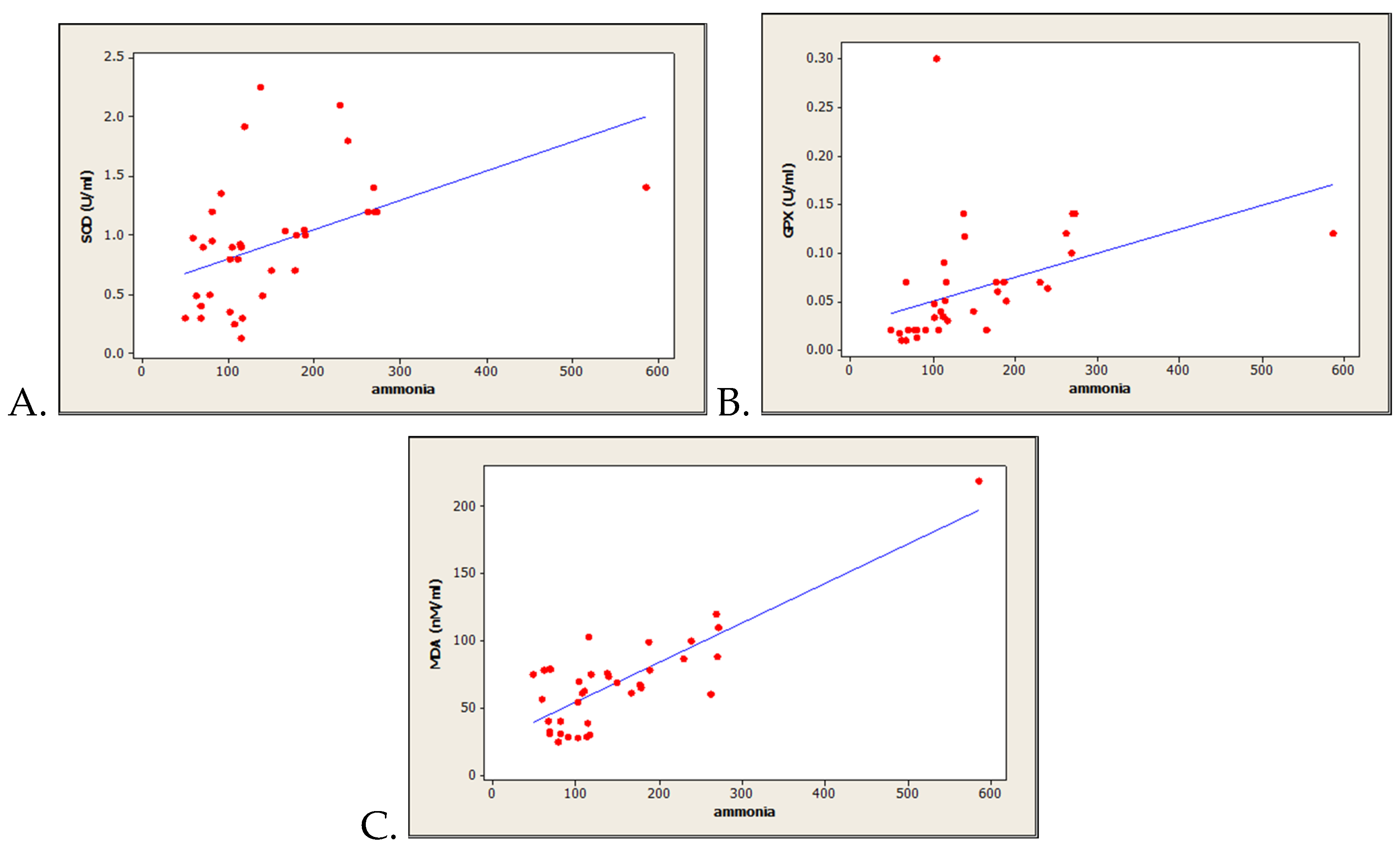
3.3. Correlations between Oxidative Stress-Related Markers and Serum Ammonia Level
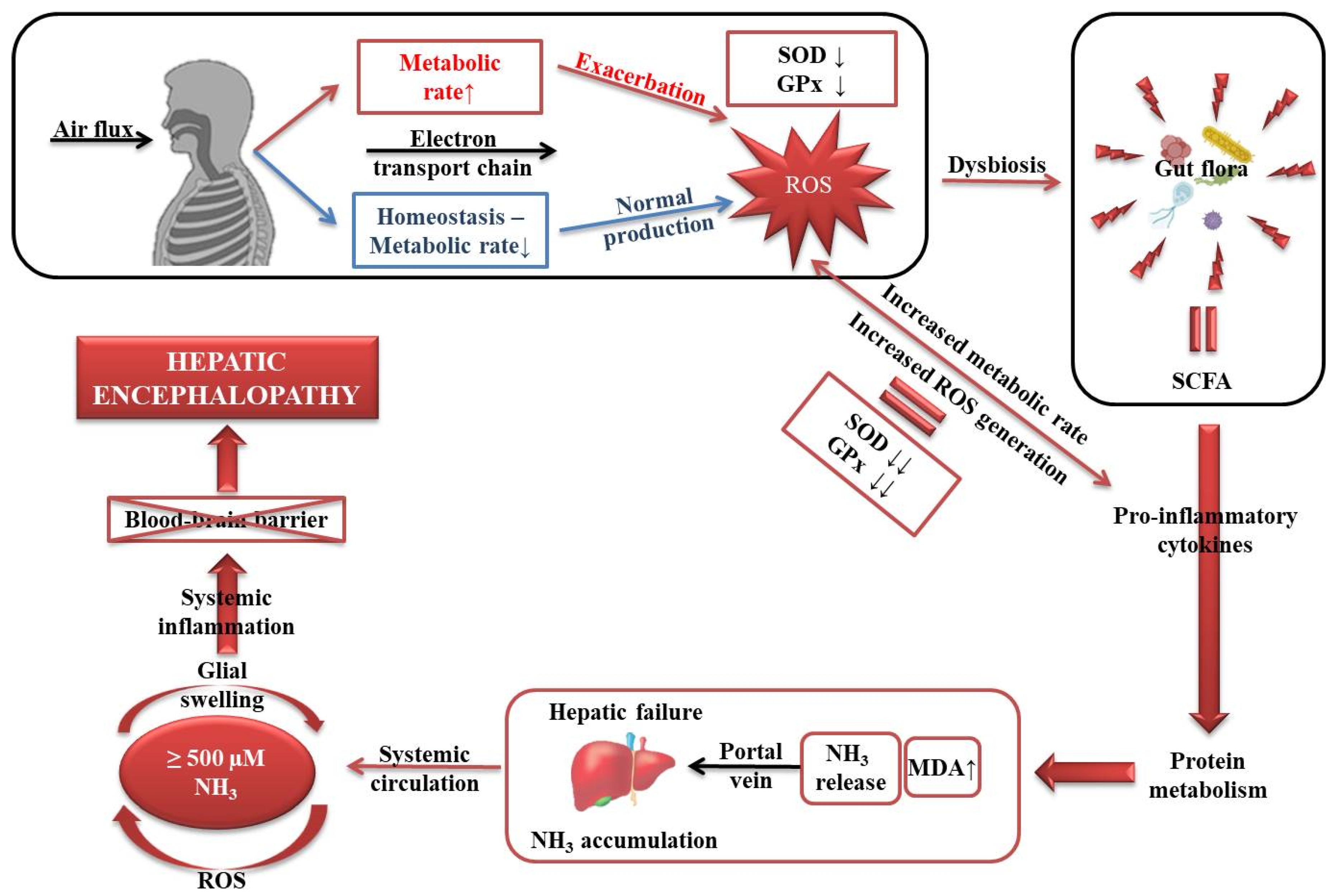
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
References
- Elwir, S.; Rahimi, R.S. Hepatic encephalopathy: An update on the pathophysiology and therapeutic options. J. Clin. Transl. Hepatol. 2017, 5, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldridge, D.R.; Tranah, E.J.; Shawcross, D.L. Pathogenesis of hepatic encephalopathy: Role of ammonia and systemic inflammation. J. Clin. Exp. Hepatol. 2015, 5, S7–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemberg, A.; Alejandra Fernández, M. Hepatic encephalopathy, ammonia, glutamate, glutamine and oxidative stress. Ann. Hepatol. 2009, 8, 95–102. [Google Scholar] [CrossRef]
- Seyan, A.S.; Hughes, R.D.; Shawcross, D.L. Changing face of hepatic encephalopathy: Role of inflammation and oxidative stress. World J. Gastroenterol. 2010, 16, 3347–3357. [Google Scholar] [CrossRef] [PubMed]
- Cichoż-Lach, H.; Michalak, A. Current pathogenetic aspects of hepatic encephalopathy and noncirrhotic hyperammonemic encephalopathy. World J. Gastroenterol. 2013, 19, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, E.; Kaminski, Y.; Lopata, O.; Muravyov, N.; Felipo, V. Blocking NMDA receptors prevents the oxidative stress induced by acute ammonia intoxication. Free Radic. Biol. Med. 1999, 26, 1369–1374. [Google Scholar] [CrossRef]
- Kosenko, E.; Kaminsky, M.; Kaminsky, A.; Valencia, M.; Lee, L.; Hermenegildo, C.; Felipo, V. Superoxide production and antioxidant enzymes in ammonia intoxication in rats. Free Radic. Res. 1997, 27, 637–644. [Google Scholar] [CrossRef]
- Kosenko, E.; Venediktova, N.; Kaminsky, Y.; Montoliu, C.; Felipo, V. Sources of oxygen radicals in brain in acute ammonia intoxication in vivo. Brain Res. 2003, 981, 193–200. [Google Scholar] [CrossRef]
- Robb, S.J.; Connor, J.R. An in vitro model for analysis of oxidative death in primary mouse astrocytes. Brain Res. 1998, 788, 125–132. [Google Scholar] [CrossRef]
- Norenberg, M.D.; Jayakumar, A.R.; Rao, K.R. Oxidative Stress in the Pathogenesis of Hepatic Encephalopathy. Metab Brain Dis. 2004, 19, 313–329. [Google Scholar] [CrossRef]
- Bertram, R.; Gram Pedersen, M.; Luciani, D.S.; Sherman, A. A simplified model for mitochondrial ATP production. J. Theor. Biol. 2006, 243, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996, 29, 169–202. [Google Scholar] [CrossRef] [PubMed]
- Schwenger, K.J.P.; Clermont-Dejean, N.; Allard, J.P. The role of the gut microbiome in chronic liver disease: The clinical evidence revised. JHEP Rep. 2019, 1, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidari, R. Brain mitochondria as potential therapeutic targets for managing hepatic encephalopathy. Life Sci. 2019, 218, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Alden, N.; Lee, K. Pathways and functions of gut microbiota metabolism impacting host physiology. Curr. Opin. Biotechnol. 2015, 36, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Blacher, E.; Elinav, E. Microbiome, metabolites and host immunity. Curr. Opin. Microbiol. 2017, 35, 8–15. [Google Scholar] [CrossRef]
- Belizário, J.E.; Faintuch, J.; Garay-Malpartida, M. Gut Microbiome dysbiosis and immunometabolism: New frontiers for treatment of metabolic diseases. Mediat. Inflamm. 2018, 2018, 2037838. [Google Scholar] [CrossRef]
- Lockwood, A.H. Blood ammonia levels and hepatic encephalopathy. Metab. Brain Dis. 2004, 19, 345–349. [Google Scholar] [CrossRef]
- Häussinger, D.; Görg, B. Interaction of oxidative stress, astrocyte swelling and cerebral ammonia toxicity. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 87–92. [Google Scholar] [CrossRef]
- Görg, B.; Qvartskhava, N.; Keitel, V.; Bidmon, H.J.; Selbach, O.; Schliess, F.; Häussinger, D. Ammonia induces RNA oxidation in cultured astrocytes and brain in vivo. Hepatology 2008, 48, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Görg, B.; Becker, S.; Qvartskhava, N.; Bidmon, H.J.; Selbach, O.; Haas, H.L.; Schliess, F.; Häussinger, D. Hypoosmotic swelling and ammonia increase oxidative stress by NADPH oxidase in cultured astrocytes and vital brain slices. Glia 2007, 55, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Irimia, R.; Ciobica, A.; Carol, S.; Trifan, A. The relevance of oxidative stress in cirrhotic patients with different forms of hepatic encephalopathy. Arch. Biol. Sci. 2013, 65, 1245–1252. [Google Scholar] [CrossRef]
- Singh, S.; Koiri, R.K.; Trigun, S.K. Acute and chronic hyperammonemia modulate antioxidant enzymes differently in cerebral cortex and cerebellum. Neurochem. Res. 2008, 33, 103–113. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef]
- Krishna Mohan, S.; Venkataramana, G. Status of lipid peroxidation, glutathione, ascorbic acid, vitamin E and antioxidant enzymes in patients with osteoarthritis. Indian J. Med. Sci. 2007, 61, 9–14. [Google Scholar]



{kind=link}
{kind=link}
| Parameter | All Patients n = 40 | Study Group A (High Ammoniemia) n = 20 | Study Group B (Normal Ammoniemia) n = 20 | P-Value |
|---|---|---|---|---|
| Gender, Male/Female (%) | 19/21 (47.5/52.5) | 11/9 (55/45) | 8/12 (40/60) | 0.342 C |
| Age, Years, Mean ± SD | 56.0 ± 10.4 | 54.3 ± 9.6 | 57.6 ± 10.4 | 0.305 T |
| Etiology of Cirrhosis, n (%) | 0.322 F | |||
| HCV | 10 (25.0) | 3 (15.0) | 7 (35) | |
| HBV | 6 (15.0) | 3 (15.0) | 3 (15.0) | |
| Alcohol | 24 (60.0) | 14 (70.0) | 10 (50.0) | |
| Child–Pugh Class B/C, n (%) | 15/25 (37.5/62.5) | 4/16 (20/80) | 11/9 (55/45) | 0.022 F |
| Child–Pugh Ccore, Median (Q1/Q3) | 10 (8/15) | 10.5 (9/15) | 9(8/13) | 0.077 M |
| MELD Score, Median (Q1/Q3) | 15 (11/20) | 21 (11/35) | 17 (8/32) | 0.356 M |
| Creatinine (mg/dL),Median (Q1/Q3) | 0.78 (0.11/3.5) | 0.85 (0.12/2.77) | 0.73 (0.11/3.5) | 0.810 M |
| Albumin (g/L), Median (Q1/Q3) | 2.8 (1.59/4.5) | 2.9 (1.5/3.5) | 2.6 (1.96/4.5) | 0.394 M |
| Bilirubin (mg/dL), Median (Q1/Q3) | 4.03 (1.74/24.6) | 2.15 (1.73/24.1) | 4.62 (1.8/16.0) | 0.121 M |
| INR, Median (Q1/Q3) | 1.5 (1.28/1.74) | 1.42 (1.22/1.63) | 1.5 (1.3/1.78) | 0.461 M |
| Ammonia (µmols/L), Median (Q1/Q3) | 117 (91.5/188) | 158 (113.5/234.5) | 69 (60.5/79.5) | 0.001 M |
| Ascites, Mild/Medium/ Large, n (%) | 10/14/16 (25/35/40) | 5/8/7 (25/40/35) | 5/6/9 (25/30/45) | 0.536 F |
| SBP, n (%) | 6 (15.0) | 2 (10.0) | 4 (20.0) | 0.096 F |
| HRS, n (%) | 5 (12.5) | 3 (15.0) | 2 (10.0) | 0.633 F |
| UGIB, n (%) | 13 (32.5) | 6 (30.0) | 7 (35.0) | 0.736 F |
| Encephalopathy, n (%) | 0.043 F | |||
| Stage II | 16 (40.0) | 9 (45.0) | 7 (35.0) | |
| Stage III | 15 (37.5) | 4 (20.0) | 11 (55.0) | |
| Stage IV | 9 (22.5) | 7 (35.0) | 2 (10.0) |
| Parameter | Controls n = 21 | All Cirrhotic Patients n = 40 | Study Group A (High Ammoniemia) n = 20 | Study Group B (Normal Ammoniemia) n = 20 |
|---|---|---|---|---|
| SOD (U/mL) | 1.35 ± 0.08 | 0.90 ± 0.08 | 1.06 ± 0.07 ª | 0.68 ± 0.08 |
| GPx (U/mL) | 0.09 ± 0.006 | 0.061 ± 0.008 | 0.08 ± 0.005 ª | 0.024 ± 0.004 |
| MDA (nmols/mL) | 35.94 ± 1.37 | 68.90 ± 5.68 | 76.93 ± 5.48 | 50.06 ± 5.60 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sfarti, C.; Ciobica, A.; Balmus, I.-M.; Ilie, O.-D.; Trifan, A.; Petrea, O.; Cojocariu, C.; Gîrleanu, I.; Sîngeap, A.M.; Stanciu, C. Systemic Oxidative Stress Markers in Cirrhotic Patients with Hepatic Encephalopathy: Possible Connections with Systemic Ammoniemia. Medicina 2020, 56, 196. https://doi.org/10.3390/medicina56040196
Sfarti C, Ciobica A, Balmus I-M, Ilie O-D, Trifan A, Petrea O, Cojocariu C, Gîrleanu I, Sîngeap AM, Stanciu C. Systemic Oxidative Stress Markers in Cirrhotic Patients with Hepatic Encephalopathy: Possible Connections with Systemic Ammoniemia. Medicina. 2020; 56(4):196. https://doi.org/10.3390/medicina56040196
Chicago/Turabian StyleSfarti, Cătălin, Alin Ciobica, Ioana-Miruna Balmus, Ovidiu-Dumitru Ilie, Anca Trifan, Oana Petrea, Camelia Cojocariu, Irina Gîrleanu, Ana Maria Sîngeap, and Carol Stanciu. 2020. "Systemic Oxidative Stress Markers in Cirrhotic Patients with Hepatic Encephalopathy: Possible Connections with Systemic Ammoniemia" Medicina 56, no. 4: 196. https://doi.org/10.3390/medicina56040196
APA StyleSfarti, C., Ciobica, A., Balmus, I. -M., Ilie, O. -D., Trifan, A., Petrea, O., Cojocariu, C., Gîrleanu, I., Sîngeap, A. M., & Stanciu, C. (2020). Systemic Oxidative Stress Markers in Cirrhotic Patients with Hepatic Encephalopathy: Possible Connections with Systemic Ammoniemia. Medicina, 56(4), 196. https://doi.org/10.3390/medicina56040196