Kidney Transplant in Fabry Disease: A Revision of the Literature


 and
and 
Abstract
:1. Introduction
2. Organ Transplantation as a Replacement Therapy for Enzyme Deficiency: An Ineffective Solution
3. Graft and Patient Outcomes in Fabry Disease Patients after Kidney Transplant
4. Enzyme Replacement Therapy Impact on Kidney Transplant Outcomes and Immunosuppressive Therapy
5. Effect of Immunosuppression on ERT
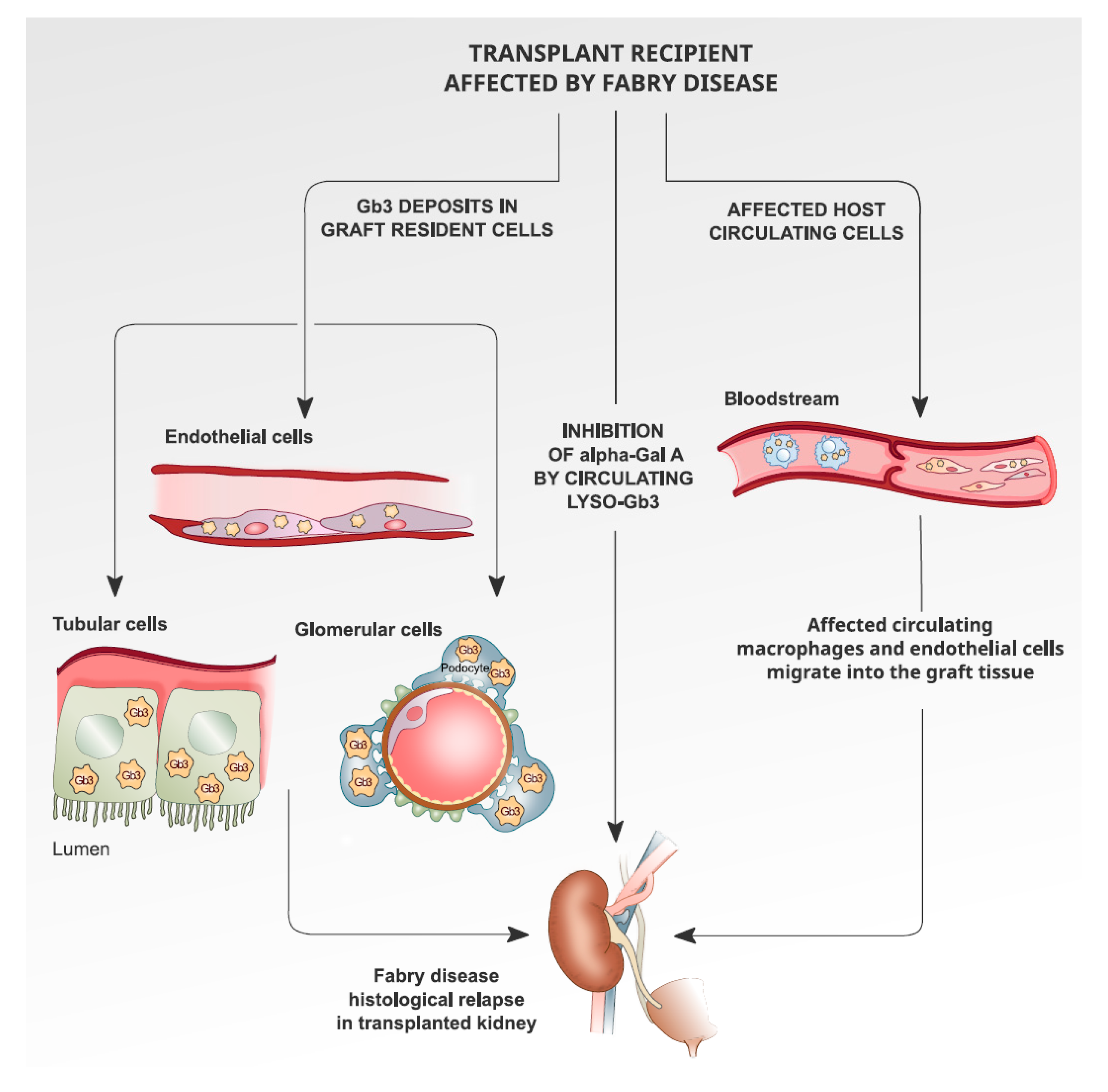
6. Recurrence of Fabry Disease in Kidney Transplantation: The Histological Findings
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; Garcia de Lorenzo, A.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur. J. Clin. Investig. 2004, 34, 236–242. [Google Scholar] [CrossRef]
- Del Pino, M.; Andrés, A.; Bernabéu, A.Á.; de Juan-Rivera, J.; Fernández, E.; de Dios García Díaz, J.; Hernández, D.; Luño, J.; Fernández, I.M.; Paniagua, J.; et al. Fabry Nephropathy: An Evidence-Based Narrative Review. Kidney Blood Press Res. 2018, 43, 296. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingelhöfer, D.; Braun, M.; Seeger-Zybok, R.K.; Quarcoo, D.; Brüggmann, D.; Groneberg, D.A. Global research on Fabry’s disease: Demands for a rare disease. Mol. Genet. Genomic Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, T.; Kido, J.; Yoshida, S.; Sugawara, K.; Momosaki, K.; Inoue, T.; Tajima, G.; Sawada, H.; Mastumoto, S.; Endo, F.; et al. Newborn screening for Fabry disease in the western region of Japan. Mol. Genet. Metab. Rep. 2020, 22, 100562. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.Y.; Chao, M.C.; Lin, S.J.; Kitagawa, T.; et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G > A (IVS4+919G > A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet. Med. 2017, 19, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Pan, X.; Ouyang, Y.; Wang, Z.; Ren, H.; Shen, P.; Wang, W.; Xu, Y.; Ni, L.; Yu, X.; Chen, X.; et al. Genotype: A Crucial but Not Unique Factor Affecting the Clinical Phenotypes in Fabry Disease. PLoS ONE 2016, 11, e0161330. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Warnock, D.G.; Banikazemi, M.; Bultas, J.; Linthorst, G.E.; Packman, S.; Sorensen, S.A.; Wilcox, W.R.; Desnick, R.J. Fabry disease: Progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol. Dial. Transplant. 2009, 24, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.; Wanner, C.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Branton, M.H.; Schiffmann, R.; Sabnis, S.G.; Murray, G.J.; Quirk, J.M.; Altarescu, G.; Goldfarb, L.; Brady, R.O.; Balow, J.E.; Austin Iii, H.A.; et al. Natural history of Fabry renal disease: Influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine 2002, 81, 122–138. [Google Scholar] [CrossRef] [PubMed]
- West, M.; Nicholls, K.; Mehta, A.; Clarke, J.T.; Steiner, R.; Beck, M.; Barshop, B.A.; Rhead, W.; Mensah, R.; Ries, M.; et al. Agalsidase Alfa and Kidney Dysfunction in Fabry Disease. J. Am. Soc. Nephrol. 2009, 20, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.V.; Granqvist, H.; Petersen, J.H.; Rasmussen, Å.K.; Lund, A.M.; Oturai, P.; Sørensen, S.S.; Feldt-Rasmussen, U. Age-related renal function decline in Fabry disease patients on enzyme replacement therapy: A longitudinal cohort study. Nephrol. Dial. Transplant. 2019, 34, 1525–1533. [Google Scholar] [CrossRef]
- Inderbitzin, D.; Avital, I.; Largiadèr, F.; Vogt, B.; Candinas, D. Kidney transplantation improves survival and is indicated in Fabry’s disease. Transplant. Proc. 2005, 37, 4211–4214. [Google Scholar] [CrossRef]
- Feriozzi, S.; Torras, J.; Cybulla, M.; Nicholls, K.; Sunder-Plassmann, G.; West, M. The effectiveness of long-term agalsidase alfa therapy in the treatment of Fabry nephropathy. Clin. J. Am. Soc. Nephrol. 2012, 7, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, B.H.; Hyang Cho, J.; Kang, E.; Choi, J.H.; Kim, G.H.; Yoo, H.W. Long-term enzyme replacement therapy for Fabry disease: Efficacy and unmet needs in cardiac and renal outcomes. J. Hum. Genet. 2016, 61, 923–929. [Google Scholar] [CrossRef]
- Philippart, M.; Franklin, S.S.; Gordon, A. Reversal of an inborn sphingolipidosis (Fabry’s disease) by kidney transplantation. Ann. Intern. Med. 1972, 77, 195–200. [Google Scholar] [CrossRef]
- Clarke, J.T.; Guttmann, R.D.; Wolfe, L.S.; Beaudoin, J.G.; Morehouse, D.D. Enzyme replacement therapy by renal allotransplantation in Fabry’s disease. N. Engl. J. Med. 1972, 287, 1215–1218. [Google Scholar] [CrossRef]
- Touraine, J.L.; Malik, M.C.; Perrot, H.; Maire, I.; Revillard, J.P.; Grosshans, E.; Traeger, J. Fabry’s disease: Two patients improved by fetal liver cells (author’s transl). Nouv. Presse Med. 1979, 8, 1499–1503. [Google Scholar]
- Likhitsup, A.; Helzberg, J.H.; Alba, L.M.; Larkin, M.K.; Cummings, L.; Island, E.R.; Lustig, R.M.; Forster, J. Persistent Alpha-galactosidase A Deficiency After Simultaneous Liver-kidney Transplantation in a Patient with Fabry Disease. Transplantation 2018, 102, e361. [Google Scholar] [CrossRef]
- Wang, R.Y.; Bodamer, O.A.; Watson, M.S.; Wilcox, W.R.; ACMG Work Group on Diagnostic Confirmation of Lysosomal Storage Diseases. Lysosomal storage diseases: Diagnostic confirmation and management of presymptomatic individuals. Genet. Med. 2011, 13, 457–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, B.A.; Bergan, J.J. Advisory Committee to the Renal Transplant Registry. Renal transplantation in Congenital and Metabolic Diseases A report from the ASC/NIH Renal Transplant Registry. JAMA 1975, 232, 148–153. [Google Scholar]
- Maizel, S.E.; Simmons, R.L.; Kjellstrand, C.; Fryd, D.S. Ten-year experience in renal transplantation for Fabry’s disease. Transplant. Proc. 1981, 13 Pt 1, 57–59. [Google Scholar]
- Tsakiris, D.; Simpson, H.K.; Jones, E.H.; Briggs, J.D.; Elinder, C.G.; Mendel, S.; Piccoli, G.; dos Santos, J.P.; Tognoni, G.; Vanrenterghem, Y.; et al. Report on management of renale failure in Europe, XXVI, 1995. Rare diseases in renal replacement therapy in the ERA-EDTA Registry. Nephrol. Dial. Transplant. 1996, 11 (Suppl. S7), 4–20. [Google Scholar] [CrossRef] [PubMed]
- Ojo, A.; Meier-Kriesche, H.U.; Friedman, G.; Hanson, J.; Cibrik, D.; Leichtman, A.; Kaplan, B. Excellent outcome of renal transplantation in patients with Fabry’s disease. Transplantation 2000, 69, 2337–2339. [Google Scholar] [CrossRef] [PubMed]
- Ersözlü, S.; Desnick, R.J.; Huynh-Do, U.; Canaan-Kühl, S.; Barbey, F.; Genitsch, V.; Mueller, T.F.; Cheetham, M.; Flammer, A.J.; Schaub, S.; et al. Long-term Outcomes of Kidney Transplantation in Fabry Disease. Transplantation 2018, 102, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Banikazemi, M.; Gordon, R.E.; Goldman, M.; Phelps, R.; Kim, L.; Gass, A.; Winston, J.; Dikman, S.; Fallon, J.T.; et al. A Phase 1/2 Clinical Trial of Enzyme Replacement in Fabry Disease: Pharmacokinetic, Substrate Clearance, and Safety Studies. Am. J. Hum. Genet. 2001, 68, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Kopp, J.B.; Austin HA 3rd Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in Fabry Disease: A randomized controlled trial. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef]
- Mignani, R.; Panichi, V.; Giudicissi, A.; Taccola, D.; Boscaro, F.; Feletti, C.; Moneti, G.; Cagnoli, L. Enzyme replacement therapy with agalsidase beta in kidney transplant patients with Fabry disease: A pilot study. Kidney Int. 2004, 65, 1381–1385. [Google Scholar] [CrossRef]
- Mignani, R.; Feriozzi, S.; Pisani, A.; Cioni, A.; Comotti, C.; Cossu, M.; Foschi, A.; Giudicissi, A.; Gotti, E.; Lozupone, V.A.; et al. Agalsidase therapy in patients with Fabry disease on renal replacement therapy: A nationwide study in Italy. Nephrol. Dial. Transplant. 2008, 23, 1628–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulla, M.; Walter, K.N.; Schwarting, A.; Divito, R.; Feriozzi, S.; Sunder-Plassmann, G.; European FOS Investigators Group. Kidney transplantation in patients with Fabry disease. Transpl. Int. 2009, 22, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo, G.; Capelli, I.; Angelini, M.L.; Valentini, C.; Baraldi, O.; Scolari, M.P.; Stefoni, S. Importance of vascular calcification in kidney transplant recipients. Am. J. Nephrol. 2014, 39, 418–426. [Google Scholar] [CrossRef] [PubMed]
- La Manna, G.; Boriani, G.; Boriani, G.; Capelli, I.; Marchetti, A.; Grandinetti, V.; Spazzoli, A.; Dalmastri, V.; Todeschini, P.; Rucci, P.; et al. Incidence and predictors of postoperative atrial fibrillation in kidney transplant recipients. Transplantation 2013, 96, 981–986. [Google Scholar] [CrossRef]
- Warnock, D.G.; Bichet, D.G.; Holida, M.; Goker-Alpan, O.; Nicholls, K.; Thomas, M.; Eyskens, F.; Shankar, S.; Adera, M.; Sitaraman, S.; et al. Oral Migalastat HCl Leads to Greater Systemic Exposure and Tissue Levels of Active α-Galactosidase A in Fabry Patients when Co-Administered with Infused Agalsidase. PLoS ONE 2015, 10, e0134341. [Google Scholar] [CrossRef]
- Müntze, J.; Gensler, D.; Maniuc, O.; Liu, D.; Cairns, T.; Oder, D.; Hu, K.; Lorenz, K.; Frantz, S.; Wanner, C.; et al. Oral Chaperone Therapy Migalastat for Treating Fabry Disease: Enzymatic Response and Serum Biomarker Changes After 1 Year. Clin. Pharmacol. Ther. 2019, 105, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Mignani, R. The management of Fabry nephropathy. Nephrol. Point Care 2016, 2, e39–e46. [Google Scholar] [CrossRef] [Green Version]
- Terryn, W.; Cochat, P.; Froissart, R.; Ortiz, A.; Pirson, Y.; Poppe, B.; Serra, A.; Van Biesen, W.; Vanholder, R.; Wanner, C. Fabry nephropathy: Indications for screening and guidance for diagnosis and treatment by the European Renal Best Practice. Nephrol. Dial. Transplant. 2013, 28, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Bénichou, B.; Goyal, S.; Sung, C.; Norfleet, A.M.; O’Brien, F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol. Genet. Metab. 2009, 96, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Oder, D.; Nowak, A.; Canaan-Kühl, S.; Arash-Kaps, L.; Drechsler, C.; Schmitz, B.; Nordbeck, P.; Hennermann, J.B.; Kampmann, C.; et al. Impact of immunosuppressive therapy on therapy-neutralizing antibodies in transplanted patients with Fabry disease. J. Intern. Med. 2017, 282, 241–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenders, M.; Stypmann, J.; Duning, T.; Schmitz, B.; Brand, S.M.; Brand, E. Serum-Mediated Inhibition of Enzyme Replacement Therapy in Fabry Disease. J. Am. Soc. Nephrol. 2016, 27, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Mosnier, J.F.; Degott, C.; Bedrossian, J.; Molas, G.; Degos, F.; Pruna, A.; Potet, F. Recurrence of Fabry’s disease in a renal allograft eleven years after successful renal transplantation. Transplantation 1991, 51, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Gantenbein, H.; Bruder, E.; Burger, H.R.; Briner, J.; Binswanger, U. Recurrence of Fabry’s disease in a renal allograft 14 years after transplantation. Nephrol. Dial. Transplant. 1995, 10, 287–289. [Google Scholar]
- Bühler, F.R.; Thiel, G.; Dubach, U.C.; Enderlin, F.; Gloor, F.; Thölen, H. Kidney Transplantation in Fabry’s Disease. Br. Med. J. 1973, 3, 28–29. [Google Scholar] [CrossRef] [Green Version]
- Faraggiana, T.; Churg, J.; Grishman, E.; Strauss, L.; Prado, A.; Bishop, D.F.; Schuchman, E.; Desnick, R.J. Light- and electron-microscopic histochemistry of Fabry’s disease. Am. J. Pathol. 1981, 103, 247–262. [Google Scholar]
- MacMahon, J.; Tubbs, R.; Gephardt, G.; Steinmuller, D. Pseudo recurrence of Fabry’s disease in renal allograft (Abstract). Lab. Investig. 1986, 54, 42A. [Google Scholar]
- Clement, M.; McGonigle, R.J.; Monkhouse, P.M.; Keogh, A.M.; Marten, R.H.; Bewick, M.; Parsons, V. Renal transplantation in Anderson-Fabry disease. J. R. Soc. Med. 1982, 75, 557–560. [Google Scholar]
- Popli, S.; Molnar, Z.V.; Leehey, D.J.; Daugirdas, J.T.; Roth, D.A.; Adams, M.B.; Cheng, J.C.; Ing, T.S. Involvement of renal allograft by Fabry’s disease. Am. J. Nephrol. 1987, 7, 316–318. [Google Scholar] [CrossRef]
- Erten, Y.; Ozdemir, F.N.; Demirhan, B.; Karakayali, H.; Demirağ, A.; Akkoç, H. A case of Fabry’s disease with normal kidney function at 10 years after successful renal transplantation. Transplant. Proc. 1998, 30, 842–843. [Google Scholar] [CrossRef]
- Friedlaender, M.M.; Kopolovic, J.; Rubinger, D.; Silver, J.; Drukker, A.; Ben-Gershon, Z.; Durst, A.L.; Popovtzer, M.M. Renal biopsy in Fabry’s disease eight years after successful renal transplantation. Clin. Nephrol. 1987, 27, 206–211. [Google Scholar] [PubMed]
- Bannwart, F. Fabry’s disease. Light and electron microscopic cardiac findings 12 years after successful kidney transplantation. Schweiz. Med. Wochenschr. 1982, 112, 1742–1747. [Google Scholar] [PubMed]


{kind=link}
| Compared Outcomes | |||||
|---|---|---|---|---|---|
| Pre ERT Era | ERT Era | Non Fabry | |||
| Ojo et al., (1988–1998) | Inderbitzin et al., (1964–1998) | Mignani et al., (2008) | Ersolozlu et al., (1979–2017) | USRDS (2006–2011) | |
| 5 years graft | 76% | 90% | 87.5% | 93% | 75% |
| 5 years patients | 83% | 100% | 100% | 100% | 85% |
| 10 years graft | 56% | 66% | 92% | 48% | |
| 10 years patients | 67% | 76% | 100% | 64% | |
| 25 years graft | 22% | ||||
| 25 years patients | 25% | ||||
| Year of Publication | Authors | Number of Patients | Biopsy Timing | Histology Findings |
|---|---|---|---|---|
| 1972 | Clarke et al. | 1 | >1 year | Glycolipids deposition |
| 1981 | Farragiana et al. | 1 | >1 year | Glomerular, tubular and interstitial deposits |
| 1973 | Buhler et al. | 1 | 5 years | No deposits |
| 1982 | Clement et al. | 1 | 5 years | No deposits |
| 1986 | McMahon et al. | 1 | 5 years | Endothelial depositions |
| 1987 | Popli et al. | 1 | 5 years | Zebra bodies |
| 1998 | Erten et al. | 1 | 5–10 years | No deposits |
| 1987 | Friedaender et al. [52] | 1 | 5–10 years | No deposits |
| 1982 | Bannwart et al. [53] | 1 | <10 years | No deposits |
| 1991 | Mosnier et al. | 1 | <10 years | No deposits |
| 1995 | Gantenbein et al. | 1 | <10 years | Tubular, interstitial deposits |
| 2018 | Ersözlü et al. | 17 | <25 years | Glomerular, tubular and interstitial deposits in 2 patients |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capelli, I.; Aiello, V.; Gasperoni, L.; Comai, G.; Corradetti, V.; Ravaioli, M.; Biagini, E.; Graziano, C.; La Manna, G. Kidney Transplant in Fabry Disease: A Revision of the Literature. Medicina 2020, 56, 284. https://doi.org/10.3390/medicina56060284
Capelli I, Aiello V, Gasperoni L, Comai G, Corradetti V, Ravaioli M, Biagini E, Graziano C, La Manna G. Kidney Transplant in Fabry Disease: A Revision of the Literature. Medicina. 2020; 56(6):284. https://doi.org/10.3390/medicina56060284
Chicago/Turabian StyleCapelli, Irene, Valeria Aiello, Lorenzo Gasperoni, Giorgia Comai, Valeria Corradetti, Matteo Ravaioli, Elena Biagini, Claudio Graziano, and Gaetano La Manna. 2020. "Kidney Transplant in Fabry Disease: A Revision of the Literature" Medicina 56, no. 6: 284. https://doi.org/10.3390/medicina56060284
APA StyleCapelli, I., Aiello, V., Gasperoni, L., Comai, G., Corradetti, V., Ravaioli, M., Biagini, E., Graziano, C., & La Manna, G. (2020). Kidney Transplant in Fabry Disease: A Revision of the Literature. Medicina, 56(6), 284. https://doi.org/10.3390/medicina56060284