
Gorham-Stout Disease with Multiple Bone Involvement—Challenging Diagnosis of a Rare Disease and Literature Review

 ,
,
Abstract
:1. Introduction
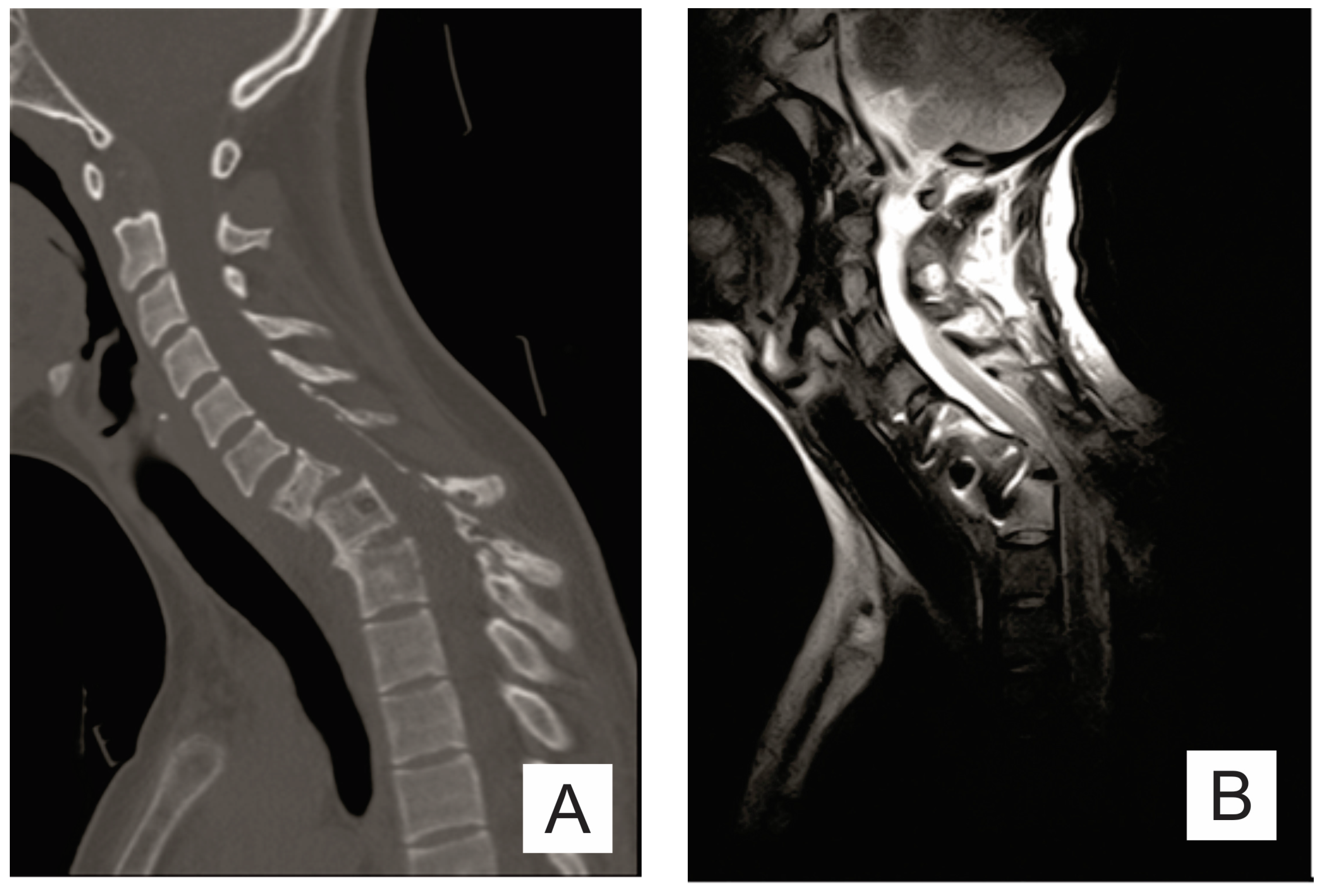
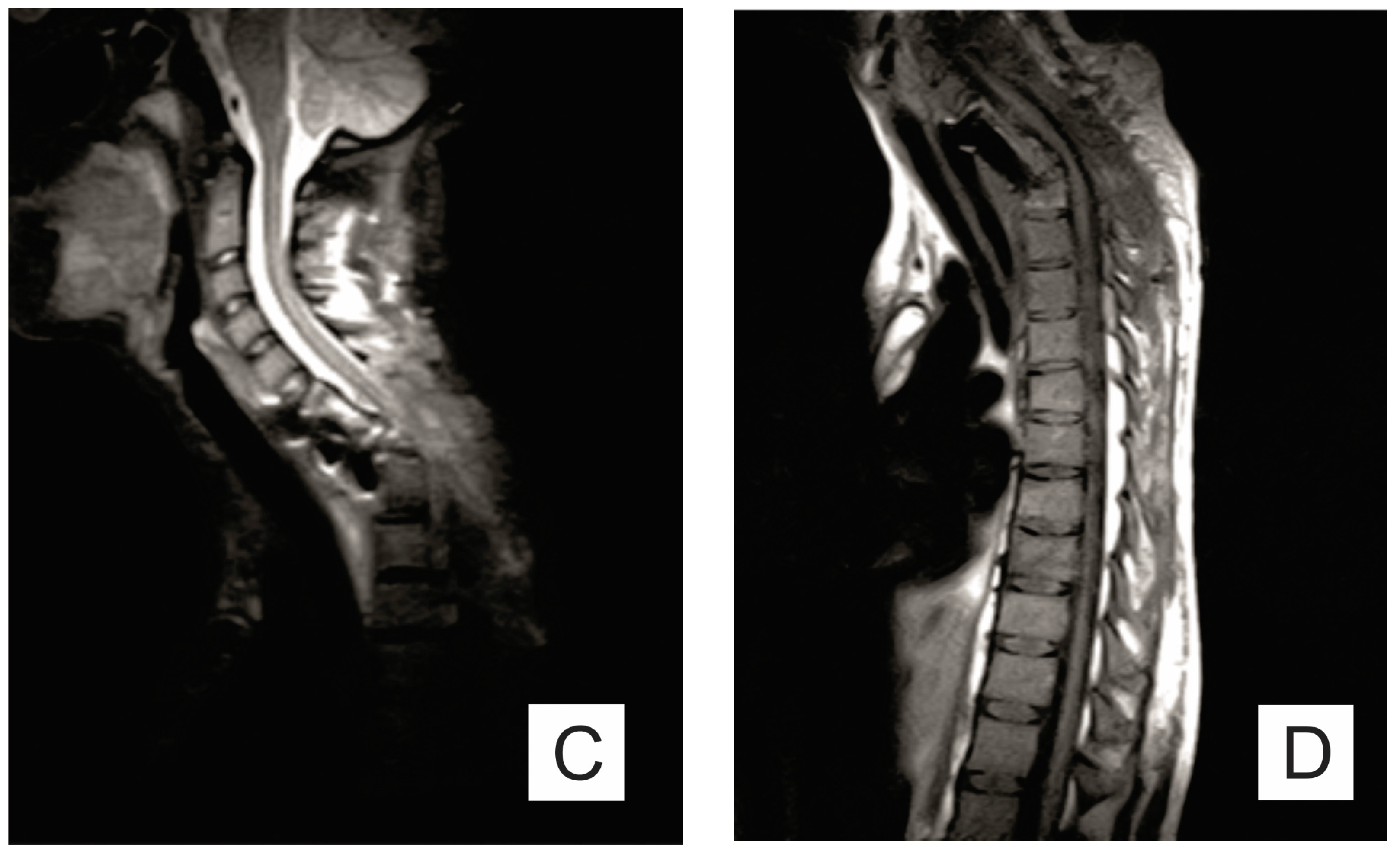
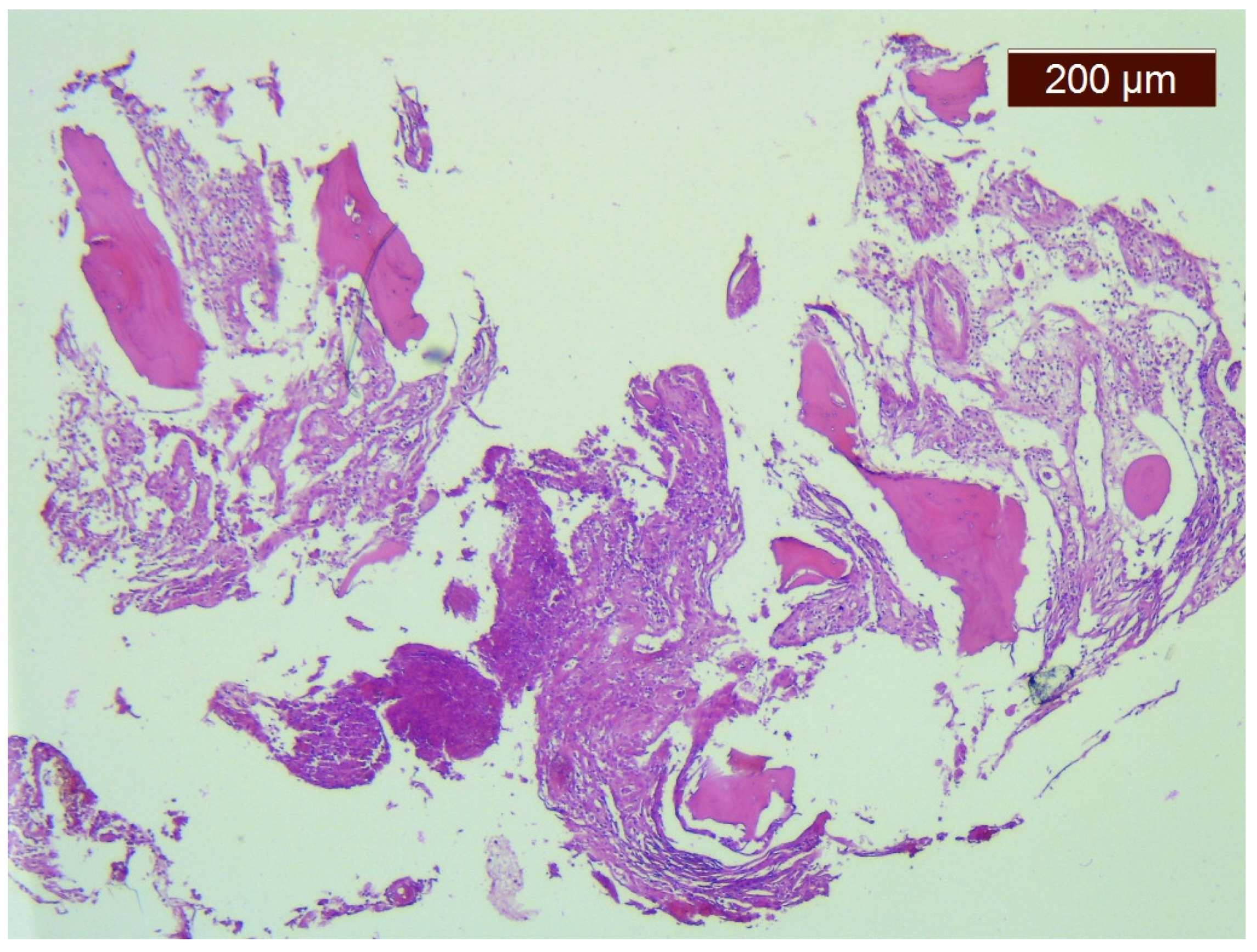
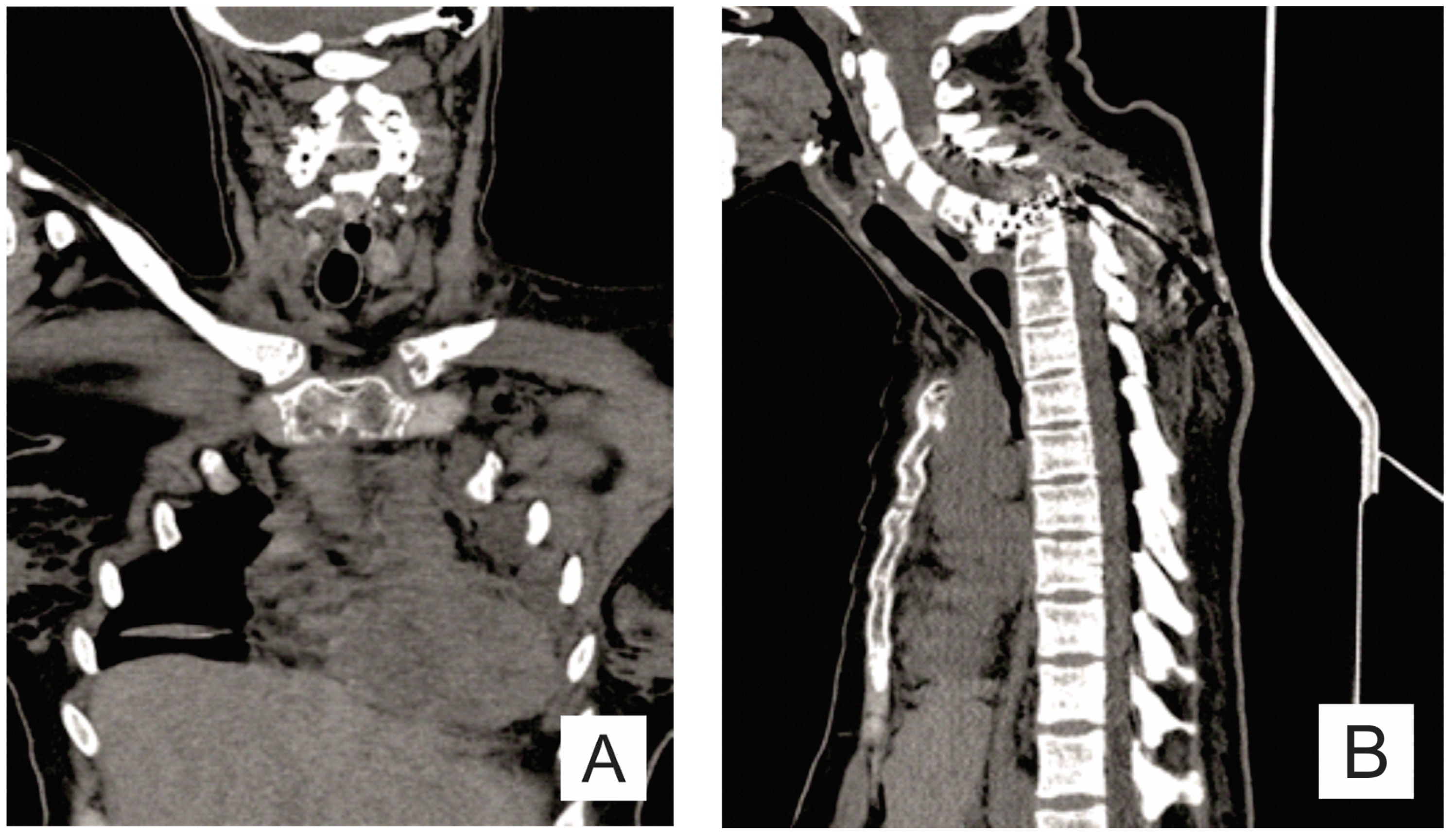
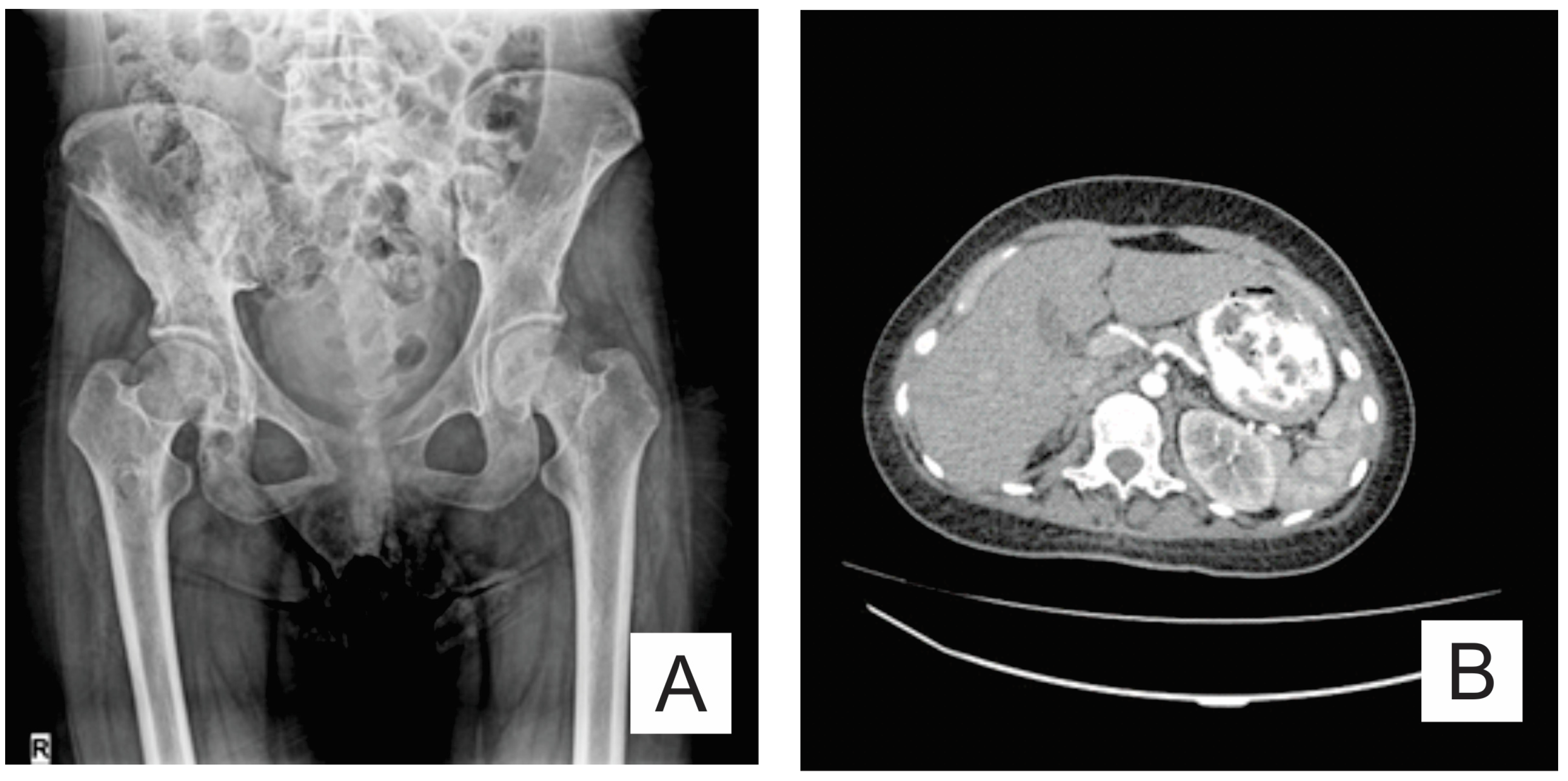
2. Case Report
3. Discussion
3.1. Clinical Features
3.2. Pathogenesis
3.3. Treatment
3.4. Life Expectancy, Complications, Mortality
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Prevalence and Incidence of Rare Diseases: Bibliographic Data. Orphanet Report Series Number 1 January 2020. Available online: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf (accessed on 5 November 2020).
- Nikolaou, V.S.; Chytas, D.; Korres, D.; Efstathopoulos, N. Vanishing bone disease (Gorham-Stout syndrome): A review of a rare entity. World J. Orthop. 2014, 5, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Hardegger, F.; Simpson, L.A.; Segmueller, G. The syndrome of idiopathic osteolysis. Classification, review, and case report. J. Bone Jt. Surg. Br. Vol. 1985, 67, 88–93. [Google Scholar] [CrossRef]
- ISSVA Classification of Vascular Anomalies ©2018 International Society for the Study of Vascular Anomalies. Available online: issva.org/classification (accessed on 23 August 2020).
- Lee, B.B.; Rockson, S.G.; Bergan, J. (Eds.) Lymphedema: A Concise Compendium of Theory and Practice; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Dellinger, M.T.; Garg, N.; Olsen, B.R. Viewpoints on vessels and vanishing bones in Gorham–Stout disease. Bone 2014, 63, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Chrcanovic, B.R.; Gomez, R.S. Gorham–Stout disease with involvement of the jaws: A systematic review. Int. J. Oral Maxillofac. Surg. 2019, 48, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Green, H.D.; Mollica, A.J.; Karuza, A.S. Gorham’s disease: A literature review and case reports. J. Foot Ankle Surg. 1995, 34, 435–441. [Google Scholar] [CrossRef]
- Commission of the European Communities. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions on Rare Diseases: Europe’s challenges. 2008. Available online: https://ec.europa.eu/health/ph_threats/non_com/docs/rare_com_en.pdf (accessed on 1 November 2020).
- Heffez, L.; Doku, H.C.; Carter, B.L.; Feeney, J.E. Perspectives on massive osteolysis: Report of a case and review of the literature. Oral Surg. Oral Med. Oral Pathol. 1983, 55, 331–343. [Google Scholar] [CrossRef]
- Kotecha, R.; Mascarenhas, L.; Jackson, H.A.; Venkatramani, R. Radiological features of Gorham’s disease. Clin. Radiol. 2012, 67, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Yu, J.S.; Resnick, D.; Vaughan, L.M.; Haghighi, P. Gorham syndrome of the thorax and cervical spine: CT and MRI findings. Skeletal Radiol. 1997, 26, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.Y.; Hong, S.W.; Chung, H.W.; Choi, J.A.; Kim, C.J.; Kang, H.S. MRI of Gorham’s disease: Findings in two cases. Skeletal Radiol. 2002, 31, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, K.N.; Shin, D.A.; Yi, S.; Kang, J.; Ha, Y. Surgical management of gorham-stout disease in cervical compression fracture with cervicothoracic fusion: Case report and review of literature. World Neurosurg. 2019, 129, 277–281. [Google Scholar] [CrossRef]
- Liu, Y.; Zhong, D.R.; Zhou, P.R.; Lv, F.; Ma, D.D.; Xia, W.B.; Li, M. Gorham-Stout disease: Radiological, histological, and clinical features of 12 cases and review of literature. Clin. Rheumatol. 2016, 35, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, W.; Qiao, C.; Han, B. Mandibular Gorham–Stout disease: A case report and literature review. Medicine 2017, 96, e8184. [Google Scholar] [CrossRef]
- Boyer, P.; Bourgeois, P.; Boyer, O.; Catonne, Y.; Saillant, G. Massive Gorham-Stout syndrome of the pelvis. Clin. Rheumatol. 2005, 24, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, P.; Montalti, M.; Angelini, A.; Alberghini, M.; Mercuri, M. Gorham–Stout disease: The experience of the Rizzoli Institute and review of the literature. Skeletal. Radiol. 2011, 40, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Du, C.Z.; Li, S.; Xu, L.; Zhou, Q.S.; Zhu, Z.Z.; Sun, X.; Qiu, Y. Spinal Gorham-Stout syndrome: Radiological changes and spinal deformities. Quant. Imaging Med. Surg. 2019, 9, 565–578. [Google Scholar] [CrossRef]
- Livesley, P.J.; Saifuddin, A.; Webb, P.J.; Mitchell, N.; Ramani, P. Gorham’s disease of the spine. Skeletal Radiol. 1996, 25, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Lala, S.; Mulliken, J.B.; Alomari, A.I.; Fishman, S.J.; Kozakewich, H.P.; Chaudry, G. Gorham-Stout disease and generalized lymphatic anomaly—clinical, radiologic, and histologic differentiation. Skeletal Radiol. 2013, 42, 917–924. [Google Scholar] [CrossRef]
- O’Callaghan, M.; Fabre, A.; McCann, J.; Healy, G.; McCarthy, A.; Keane, M.P.; McDonnell, T.J.; McCarthy, C. A 34-Year-Old Man With a Chylothorax and Bony Pain. Chest 2020, 157, e131–e136. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, M.; Fujino, A.; Matsuoka, K.; Nosaka, S.; Kuroda, T.; Fukao, T. Clinical features and prognosis of generalized lymphatic anomaly, Kaposiform Lymphangiomatosis, and Gorham-Stout disease. Pediatr Blood Cancer. 2016, 63, 832–838. [Google Scholar] [CrossRef]
- Wassef, M.; Blei, F.; Adams, D.; Alomari, A.; Baselga, E.; Berenstein, A.; Burrows, P.; Frieden, I.J.; Garzon, M.C.; Lopez-Gutierrez, J.C.; et al. Vascular anomalies classification: Recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics. 2015, 136, e203–e214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, M.Y.; Fares, J.; Fares, Y.; Abboud, J.A. Gorham-stout disease of the shoulder: Clinical, pathologic and therapeutic considerations. Arch. Bone Jt. Surg. 2020, 8, 58–69. [Google Scholar] [CrossRef]
- Rossi, M.; Buonuomo, P.S.; Battafarano, G.; Conforti, A.; Mariani, E.; Algeri, M.; Del Fattore, A. Dissecting the mechanisms of bone loss in Gorham-Stout disease. Bone. 2020, 130, 115068. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Rana, I.; Buonuomo, P.S.; Battafarano, G.; Mariani, E.; D’Agostini, M.; Del Fattore, A. Dysregulated miRNAs in bone cells of patients with Gorham-Stout disease. FASEB J. 2021, 35, e21424. [Google Scholar] [CrossRef]
- Colucci, S.; Taraboletti, G.; Primo, L.; Viale, A.; Roca, C.; Valdembri, D.; Geuna, M.; Pagano, M.; Grano, M.; Pogrel, A.M.; et al. Gorham-Stout syndrome: A monocyte-mediated cytokine propelled disease. J. Bone Min. Res. 2006, 21, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Bode-Lesniewska, B.; Von Hochstetter, A.; Exner, G.; Hodler, J. Gorham-Stout disease of the shoulder girdle and cervico-thoracic spine: Fatal course in a 65-year-old woman. Skelet. Radiol. 2002, 31724–31729. [Google Scholar] [CrossRef]
- Gondivkar, S.M.; Gadbail, A.R. Gorham-Stout syndrome: A rare clinical entity and review of literature. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2010, 109, e41–e48. [Google Scholar] [CrossRef] [PubMed]
- Moller, G.; Priemel, M.; Amling, M.; Werner, M.; Kuhlmey, A.S.; Delling, G. The Gorham–Stout syndrome (Gorham’smassive osteolysis). A report of six cases with histopathological findings. J. Bone Jt. Surg Br. 1999, 81, 501–506. [Google Scholar] [CrossRef]
- Kawasaki, K.; Ito, T.; Tsuchiya, T.; Takahashi, H. Is angiomatosis an intrinsic pathohistologi- cal feature of massive osteolysis? Report of an autopsy case and a review of the literature. Virchows Arch. 2003, 442, 400–406. [Google Scholar] [CrossRef]
- Rauh, G.; Gross, M. Disappearing bone disease (Gorham-stout disease): Report of a case with a follow-up of 48 years. Eur. J. Med Res. 1997, 2, 425–427. [Google Scholar]
- Ellati, R.; Attili, A.; Haddad, H.; Al-Hussaini, M.; Shehadeh, A.J.E.R.M.P.S. Novel approach of treating Gorham-Stout disease in the humerus–case report and review of literature. Eur. Rev. Med. Pharm. Sci. 2016, 20, 426–432. [Google Scholar]
- Ozeki, M.; Nozawa, A.; Yasue, S.; Endo, S.; Asada, R.; Hashimoto, H.; Fukao, T. The impact of sirolimus therapy on lesion size, clinical symptoms, and quality of life of patients with lymphatic anomalies. Orphanet J. Rare Dis. 2019, 14, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, H.R.; Chan, D.P.K.; Lee, J. Massive osteolysis of the cervical spine a case report of bone graft failure. Spine 1981, 6, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Yuan, X.G.; Hu, X.Y.; Shen, F.R.; Wang, J.A. Gorham-Stout syndrome in mainland China: A case series of 67 patients and review of the literature. J. Zhejiang Univ. Sci. B 2013, 14, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyd, R.; Micke, O.; Surholt, C.; Berger, B.; Martini, C.; Füller, J.; Schimpke, T.; Seegenschmiedt, M.H. Radiation therapy for Gorham-Stout syndrome: Results of a national patterns-of-care study and literature review. Int. J. Rad. Oncol. Biol. Phys. 2011, 81, e179–185. [Google Scholar] [CrossRef] [PubMed]
- Turra, S.; Gigante, C.; Scapinelli, R. A twenty-year followup study of a case of surgically treated massive osteolysis. Clin. Orthop. 1990, 250, 297–302. [Google Scholar]
- Shimizu, T.; Sato, K.; Yoshida, T.; Takahashi, A.; Yanagawa, T.; Wada, N.; Watanabe, H. A case report of Gorham–Stout syndrome remission. J. Orthop. Sci. 2012, 17, 199–204. [Google Scholar] [CrossRef]
- Lee, S.; Finn, L.; Sze, R.W.; Perkins, J.A.; Sie, K.C. Gorham Stout syndrome (disappearing bone disease): Two additional case reports and a review of the literature. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 1340–1343. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laboratory Test | Reference Value | Patient Data | Laboratory Test | Reference Value | Patient Data |
|---|---|---|---|---|---|
| Calcium | 4.61–5.17 mg/dL | 4.98 | CA 15-3 | 0–32.4 U/mL | 12.4 |
| Magnesium | 1.6–2.5 mg/dL | 1.94 | CA 19-9 | 0–30.9 IU/mL | 7.29 |
| PTH | 15–65 pg/mL | 18.22 | CEA | 0–2.5 ng/mL | <0.01 |
| Procalcitonin | 0.05–0.5 ng/mL | <0.12 | CA 125 | 1.9–16.3 U/mL | 11.3 |
| 25-hydroxy vitamin D | 30–100 ng/mL | 15.4 | CK-MB | 0–24 U/L | 8.92 |
| β−CTX | ≤0.573 ng/mL | 0.68 | Rheumatoid factor | <14 IU/mL | 6 |
| BSAP | 3–19 μg/L | 11.6 | Antithyroglobulin antibody | ≤115 IU/mL | 13.58 |
| Osteocalcin | 11–43 ng/mL | 13 | Anti-dsDNA antibody | <100 IU/mL | <10 |
| Alkaline phosphatase | 204.59 U/L | 0–800 U/L | Antinuclear antibody | <1/80 | <1/80 |
| Fibrinogen | 200–400 mg/dL | 527 mg/dl | CIC | <20 RU/mL | 5.404 |
| ESR | 2–20 mm/hour | 80 | ACE | 12–68 U/L | 45 |
| CRP | 0.1–4.1 mg/L | 61.8 | CAL | <9.82 pg/mL | 0.528 |
| Time | Surgical Treatment |
|---|---|
| Month I | C7 corpectomy and reconstruction with autologous graft from the iliac crest, macroscopic resection of the posterior arches C7-T1 and fixation with C4-C5-T3-T4 cervico-thoracic hybrid system |
| Month III | Ablation of osteosynthesis material and bone graft and T1 corpectomy, reconstruction with C6-T1 mesh fixed with proximal and distal screw |
| Month IX | Ablation of damaged osteosynthesis material mesh C6-T1, ablation of thoracic screws, and introduction of bilateral T3, T4, and T5 screws |
| Time | Neurological Deficits | Neuromotor Recovery Treatment |
|---|---|---|
| Month IV | Lower limb motor deficit (MRC 1/5 proximal and 0/5 distal), bilateral pyramidal syndrome (elastic hypertonia Ashworth 3, brisk DTR, absent SAR, positive Babinski sign bilateral, left Achilles tendon clonus), and inability to walk, but possible orthostatism for a short time with assisted support | physiotherapy, massage, kinetotherapy, pentoxifylline, gabapentin, baclofen, alfacalcidol, vitamin D and calcium supplements |
| Month VII | ||
| Month XII | Lower limb motor deficit (MRC 1/5 proximal and 0/5 distal), bilateral pyramidal syndrome (elastic hypertonia Ashworth 3, brisk DTR, absent SAR, positive Babinski sign bilateral, left Achilles tendon clonus), and inability to stand and walk |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Momanu, A.; Caba, L.; Gorduza, N.C.; Arhire, O.E.; Popa, A.D.; Ianole, V.; Gorduza, E.V. Gorham-Stout Disease with Multiple Bone Involvement—Challenging Diagnosis of a Rare Disease and Literature Review. Medicina 2021, 57, 681. https://doi.org/10.3390/medicina57070681
Momanu A, Caba L, Gorduza NC, Arhire OE, Popa AD, Ianole V, Gorduza EV. Gorham-Stout Disease with Multiple Bone Involvement—Challenging Diagnosis of a Rare Disease and Literature Review. Medicina. 2021; 57(7):681. https://doi.org/10.3390/medicina57070681
Chicago/Turabian StyleMomanu, Alina, Lavinia Caba, Nicoleta Carmen Gorduza, Oana Elena Arhire, Alina Delia Popa, Victor Ianole, and Eusebiu Vlad Gorduza. 2021. "Gorham-Stout Disease with Multiple Bone Involvement—Challenging Diagnosis of a Rare Disease and Literature Review" Medicina 57, no. 7: 681. https://doi.org/10.3390/medicina57070681
APA StyleMomanu, A., Caba, L., Gorduza, N. C., Arhire, O. E., Popa, A. D., Ianole, V., & Gorduza, E. V. (2021). Gorham-Stout Disease with Multiple Bone Involvement—Challenging Diagnosis of a Rare Disease and Literature Review. Medicina, 57(7), 681. https://doi.org/10.3390/medicina57070681