Novel Marine Phenazines as Potential Cancer Chemopreventive and Anti-Inflammatory Agents

Abstract
:
1. Introduction
2. Results and Discussion
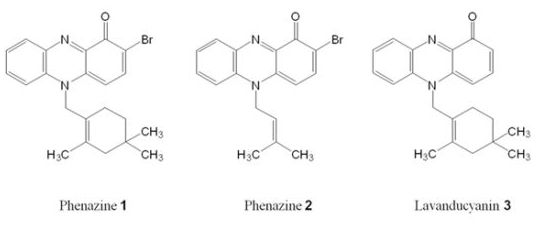
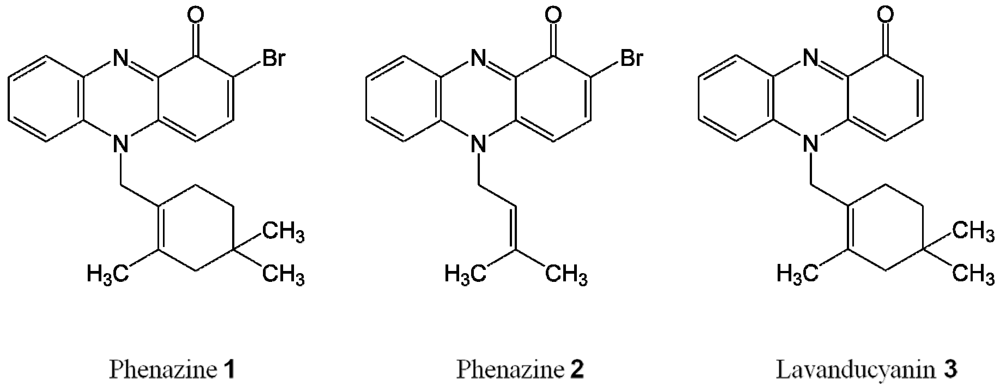
2.1. Phenazines Isolation

2.2. Phenazines Inhibit TNF-α-Induced NFκB Activity
2.3. Transcriptional Regulation of iNOS and COX-2 by Phenazines

2.4. Inhibition of NO and PGE2 Production in LPS-Induced RAW 264.7 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | NO | NO | NO | PGE2 | PGE2 |
|---|---|---|---|---|---|
| % inhibition (50 µM) | % cell survival | IC50, µM | % inhibition (50 µM) | IC50, µM | |
| Phenazine 1 | 40.1 ± 5 | 68.9 ± 3.8 | >48.6 | 72.5 ± 16.3 | 7.15 ± 2.03 |
| Phenazine 2 | 98.1 ± 0.5 | 56.0 ± 5.1 | 15.1 ± 2.7 | 86.9 ± 12.9 | 0.89 ± 0.22 |
| Lavanducyanin 3 | 97 ± 0.7 | 58.3 ± 4.6 | 8.0 ± 0.39 | 80.6 ± 18.4 | 0.63 ± 0.16 |
| Resveratrol | 93.0 ± 0.8 | 86.9 ± 3.9 | 31.9 ± 1.8 | 70.2 ± 13.9 | 2.5 ± 0.43 |
2.5. Direct Inhibition of COX-1 and COX-2 Activity
| COX-1, IC50, µM | COX-2, IC50, µM | Selectivity COX-1/COX-2 | |
|---|---|---|---|
| Phenazine 1 | 11.0 ± 0.53 | 4.0 ± 0.41 | 2.75 |
| Phenazine 2 | 5.6 ± 0.61 | 7.2 ± 0.13 | 0.78 |
| Lavanducyanin 3 | 30.0 ± 1.08 | 34.0 ± 1.1 | 0.88 |
| Indomethacin | 0.42 ± 0.21 | ND | |
| Celecoxib | ND | 0.05 ± 0.03 |
2.6. Phenazines Induce Apoptosis

3. Experimental Section
3.1. Cell Lines and Reagents
3.2. NFκB Assay
3.3. Nitric Oxide (NO) Synthase Assay
3.4. Cell Viability Assay
3.5. Determination of PGE2
3.6. COX-1 and COX-2 Inhibition Assays
3.7. Cell Cycle Analysis
3.8. Detection of Apoptosis
3.9. Real Time Quantitative PCR
3.10. Evaluation of Antioxidant Capacity
3.11. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Tergaonkar, V. NFκB pathway: A good signaling paradigm and therapeutic target. Intern. J. Biochem. Cell Biol. 2006, 38, 1647–1653. [Google Scholar]
- Aggarwal, B.B.; Sethi, G.; Nair, A.; Ichikawa, H. Nuclear Factor-κB a holy grail in cancer prevention and therapy. Curr. Sign. Transd. Ther. 2006, 1, 25–52. [Google Scholar]
- Melisi, D.; Chiao, P.J. NF-κB as a target for cancer therapy. Expert Opin. Ther. Targets 2007, 11, 133–144. [Google Scholar]
- Kim, H.J.; Hawke, N.; Baldwin, A.S. NF-κB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006, 13, 738–747. [Google Scholar]
- Pikarsky, E.; Ben-Neriah, Y. NF-κB inhibition: A double-edged sword in cancer? Eur. J. Cancer 2006, 42, 779–784. [Google Scholar] [CrossRef]
- Luqman, S.; Pezzuto, J.M. NFκB: A promising target for natural products in cancer chemoprevention. Phytother. Res. 2010, 24, 949–963. [Google Scholar]
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J. Clin. Invest. 2001, 107, 135–142. [Google Scholar]
- Gonda, T.A.; Tu, S.; Wang, T.C. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle 2009, 8, 2005–2013. [Google Scholar]
- Murakami, A. Chemoprevention with phytochemicals targeting inducible nitric oxide synthase. Forum Nutr. 2009, 61, 193–203. [Google Scholar]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar]
- Inoue, J.; Gohda, J.; Akiyama, T.; Semba, K. NF-κB activation in development and progression of cancer. Cancer Sci. 2007, 98, 268–274. [Google Scholar]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar]
- Surh, Y.J.; Kundu, J.K. Cancer preventive phytochemicals as speed breakers in inflammatory signaling involved in aberrant COX-2 expression. Curr. Cancer Drug Targets 2007, 7, 447–458. [Google Scholar]
- Kundu, J.K.; Surh, Y.J. Inflammation: Gearing the journey to cancer. Mutat. Res. 2008, 659, 15–30. [Google Scholar]
- Cuendet, M.; Pezzuto, J.M. The role of cyclooxygenase and lipoxygenase in cancer chemoprevention. Drug Metabol. Drug Interact. 2001, 17, 109–157. [Google Scholar]
- Mann, J.R.; DuBois, R.N. Cyclooxygenase-2 and gastrointestinal cancer. Cancer J. 2004, 10, 145–152. [Google Scholar]
- King, K.L.; Cidlowski, J.A. Cell cycle regulation and apoptosis. Annu. Rev. Physiol. 1998, 60, 601–617. [Google Scholar]
- Kerr, J.F.; Winterford, C.M.; Harmon, B.V. Apoptosis. Its significance in cancer and cancer therapy. Cancer 1994, 73, 2013–2026. [Google Scholar] [CrossRef]
- Kam, P.C.A.; Ferch, N.I. Apoptosis: Mechanisms and clinical implications. Anesthesia 2000, 55, 1081–1093. [Google Scholar]
- Price-Whelan, A.; Dietrich, L.E.; Newman, D.K. Rethinking “secondary” metabolism: Physiological roles for phenazine antibiotics. Nat. Chem. Biol. 2006, 2, 71–78. [Google Scholar]
- Li, D.; Wang, F.; Xiao, X.; Zeng, X.; Gu, Q.Q.; Zhu, W. A new cytotoxic phenazine derivative from a deep sea bacterium Bacillus sp. Arch. Pharm. Res. 2007, 30, 552–555. [Google Scholar]
- Mitova, M.I.; Lang, G.; Wiese, J.; Imhoff, J.F. Subinhibitory concentrations of antibiotics induce phenazine production in a marine Streptomyces sp. J. Nat. Prod. 2008, 71, 824–827. [Google Scholar]
- Conda-Sheridan, M.; Marler, L.; Park, E.-J.; Kondratyuk, T.P.; Jermihov, K.; Mesecar, A.D.; Pezzuto, J.M.; Asolkar, R.N.; Fenical, W.; Cushman, M. Potential chemopreventive agents based on the structure of the lead compound 2-bromo-1-hydroxyphenazine, isolated from Streptomyces species, strain CNS284. J. Med. Chem. 2010, 53, 8688–8699. [Google Scholar]
- Laursen, J.; Nielsen, J. Phenazine natural products: Biosynthesis, synthetic analogues, and biological activity. Chem. Rev. 2004, 104, 1663–1685. [Google Scholar]
- Kerr, J.R. Phenazine pigments: Antibiotics and virulence factors. Infect. Dis. Rev. 2000, 2, 184–194. [Google Scholar]
- Shin-Ya, K.; Shimizu, S.; Kunigami, T.; Furihata, T.; Hayakawa, Y.; Seto, H. Novel neuronal cell protecting substances, aestivophoenins A and B, produced by Streptomyces purpeofuscus. J. Antibiot. 1995, 48, 1378–1381. [Google Scholar]
- Marler, L.; Conda-Sheridan, M.; Cinelli, M.A.; Morrell, A.E.; Cushman, M.; Chen, L.; Huang, K.; van Breemen, R.B.; Pezzuto, J.M. Cancer chemopreventive potential of aromathecins and phenazines, novel natural product derivatives. Anticancer Res. 2010, 30, 4873–4882. [Google Scholar]
- Imai, S.; Furihata, K.; Hayakawa, Y.; Noguchi, T.; Seto, H. Lavanducyanin, a new antitumor substance produced by Streptomyces sp. J. Antibiot. 1989, 42, 1196–1198. [Google Scholar]
- Imai, S.; Noguchi, T.; Seto, H. Studies on cell growth stimulating substances of low molecular weight. Part 2. Exfoliazone and lavanducyanin, potent growth promoting substances of rat liver cell line, RLN-8, produced by Streptomyces exfoliatus and Streptomyces aeriouvifer. J. Antibiot. 1993, 46, 1232–1238. [Google Scholar]
- Matsumoto, M.; Seto, H. Stimulation of mammalian cell proliferation by lavanducyanin. J. Antibiot. 1991, 44, 1471–1473. [Google Scholar]
- Nakayama, O.; Arakawa, H.; Yagi, M.; Tanaka, M.; Kiyoto, S.; Okuhara, M.; Kohsaka, M. WS-9659 A and B, novel testosterone 5 alpha-reductase inhibitors isolated from a Streptomyces. III. Biological characteristics and pharmacological characteristics. J. Antibiot. 1989, 42, 1235–1240. [Google Scholar]
- Asolkar, R.; Jensen, P.R.; Fenical, W. Marinophenazines, brominated antitumor-antibiotics from a marine bacterium related to the genus Streptomyces. J. Nat. Prod. submitted for publication.
- Maiti, A.; Cuendet, M.; Kondratyuk, T.; Croy, V.L.; Pezzuto, J.M.; Cushman, M. Synthesis and cancer chemopreventive activity of zapotin, a natural product from Casimiroa edulis. J. Med. Chem. 2007, 50, 350–355. [Google Scholar]
- Asolkar, R.N.; Freel, K.C.; Jensen, P.R.; Fenical, W.; Kondratyuk, T.P.; Park, E.J.; Pezzuto, J.M. Arenamides A–C, cytotoxic NF-κB inhibitors from the marine actinomycete Salinispora arenicola. J. Nat. Prod. 2009, 72, 396–402. [Google Scholar]
- Choi, I.K.; Shin, H.J.; Lee, H.S.; Kwon, H.J. Streptochlorin, a marine natural product, inhibits NF-κB activation and suppresses angiogenesis in vitro. J Microb. Biotechnol. 2007, 17, 1338–1343. [Google Scholar]
- Folmer, F.; Jaspars, M.; Dicato, M.; Diederich, M. Marine natural products as targeted modulators of the transcription factor NF-κB. Biochem. Pharmacol. 2008, 75, 603–617. [Google Scholar]
- Ohsugi, T.; Horie1, R.; Kumasaka, T.; Ishida, A.; Ishida, T.; Yamaguchi, K.; Watanabe, T.; Umezawa, K.; Urano, T. In vivo antitumor activity of the NF-kB inhibitor dehydroxymethylepoxyquinomicin in a mouse model of adult T-cell leukemia. Carcinogenesis 2005, 26, 1382–1388. [Google Scholar]
- Chopra, P.; Bajpai, M.; Dastidar, S.G.; Ray, A. Development of a cell death-based method for the screening of nuclear factor-κB inhibitors. J. Immunol. Meth. 2008, 335, 126–131. [Google Scholar]
- Frydrych, I.; Mlejnek, P. Serine protease inhibitors N-tosyl-L-lysinyl-chloromethylketone (TLCK) and N-tosyl-L-phenylalaninyl-chloromethylketone (TPCK) are potent inhibitors of activated caspase proteases. J. Cell. Biochem. 2008, 103, 1646–1656. [Google Scholar]
- García, M.G.; Alaniz, L.; Lopes, E.C.; Blanco, G.; Hajos, S.E.; Alvarez, E. Inhibition of NF-κB activity by BAY 11-7082 increases apoptosis in multidrug resistant leukemic T-cell lines. Leuk. Res. 2005, 29, 1425–1434. [Google Scholar]
- Chung, H.T.; Choi, B.M.; Kwon, Y.G.; Kim, Y.M. Interactive relations between nitric oxide (NO) and carbon monoxide (CO): Heme oxygenase-1/CO pathway is a key modulator in NO-mediated antiapoptosis and anti-inflammation. Meth. Enzymol. 2008, 441, 329–338. [Google Scholar]
- Hofseth, L.J.; Hussain, S.P.; Wogan, G.N.; Harris, C.C. Nitric oxide in cancer and chemoprevention. Free Rad. Biol. Med. 2003, 34, 955–968. [Google Scholar]
- Bhat, K.P.; Pezzuto, J.M. Cancer chemopreventive activity of resveratrol. Ann. NY Acad. Sci. 2002, 957, 210–229. [Google Scholar]
- Santini, M.T.; Rainaldi, G.; Indovina, P.L. Apoptosis, cell adhesion and the extracellular matrix in the three-dimensional growth of multicellular tumor spheroids. Crit. Rev. Oncol. Hematol. 2000, 36, 75–87. [Google Scholar]
- Weigert, A.; Jennewein, C.; Brune, B. The liaison between apoptotic cells and macrophages—the end programs the beginning. Biol. Chem. 2009, 390, 379–390. [Google Scholar]
- Hoshino, J.; Park, E.J.; Kondratyuk, T.P.; Marler, L.; Pezzuto, J.M.; van Breemen, R.B.; Mo, S.; Li, Y.; Cushman, M. Selective synthesis and biological evaluation of sulfate-conjugated resveratrol metabolites. J. Med. Chem. 2010, 53, 5033–5043. [Google Scholar]
- Kim, Y.; Min, H.Y.; Park, H.J.; Lee, E.J.; Park, E.J.; Hwang, H.J.; Jin, C.; Lee, Y.S.; Lee, S.K. Suppressive effects of nitric oxide production and inducible nitric oxide synthase (iNOS) gene expression by Calystegia soldanella methanol extract on lipopolysaccharide-activated RAW 264.7 cells. Eur. J. Cancer Prev. 2004, 13, 419–424. [Google Scholar] [CrossRef]
- Park, E.-J.; Cheenpracha, S.; Chang, L.C.; Kondratyuk, T.P.; Pezzuto, J.M. Inhibition of lipopolysaccharide-induced cyclooxygenase-2 and inducible nitric oxide synthase expression by 4-[(2'-O-acetyl-α-L-rhamnosyloxy)benzyl]isothiocyanate from Moringa oleifera. Nutr. Cancer 2011, 63, 971–982. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bodesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Nat. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar]
- You, M.; Wickramaratne, D.B.; Silva, G.L.; Chai, H.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Kinghorn, A.D.; Pezzuto, J.M. Roemerine, an aporphine alkaloid from Annona senegalensis that reverses the multidrug-resistance phenotype with cultured cells. J. Nat. Prod. 1995, 58, 598–604. [Google Scholar]
- Mi, Q.; Cui, B.; Silva, G.L.; Lantvit, D.; Lim, E.; Chai, H.; You, M.; Hollingshead, M.G.; Mayo, J.G.; Kinghorn, A.D.; et al. Pervilleine A, a novel tropane alkaloid that reverses the multidrug-resistance phenotype. Cancer Res. 2001, 61, 4030–4037. [Google Scholar]
- Cuendet, M.; Mesecar, A.D.; DeWitt, D.L.; Pezzuto, J.M. An ELISA method to measure inhibition of the COX enzymes. Nat. Protoc. 2006, 1, 1915–1921. [Google Scholar]
- Cao, H.; Yu, R.; Tao, Y.; Nikolic, D.; van Breemen, R.B. Measurement of cyclooxygenase inhibition using liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2011, 54, 230–235. [Google Scholar]
- Yu, R.; Xiao, L.; Zhao, G.; Christman, J.W.; van Breemen, R.B. Competitive enzymatic interactions determine the relative amounts of prostaglandins E2 and D2. J. Pharmacol. Exp. Ther. 2011, 339, 716–725. [Google Scholar]
- Prism 5 for Mac OSX, version 5.0c, GraphPad Software, Inc.: Mountain View, CA, USA, 2009.
- Krishna, A.; Cabana, R. Flow cytometric analysis of electronic nuclear volume and DNA content in normal mouse tissues. Cell Cycle 2004, 3, 380–383. [Google Scholar]
- Li, X.; Darzynkiewicz, Z. Labeling DNA strand breaks with BrdUTP. Detection of apoptosis and cell proliferation. Cell Prolif. 1995, 28, 572–579. [Google Scholar]
- Zhao, L.; Tao, J.-Y.; Zhang, S.-L.; Jin, F.; Pang, R.; Dong, J.-H. N-butanol extract from Melilotus Suaveolens Ledeb affects pro- and anti-inflammatory cytokines and mediators. Evid. Based Complement Alternat. Med. 2010, 7, 97–106. [Google Scholar]
- Tsai, S.-H.; Lin-Shiau, S.-Y.; Lin, J.-K. Suppression of nitric oxide synthase and the down-regulation of the actvation of NFκB in macrophages by resveratrol. Br. J. Pharm. 1999, 126, 673–680. [Google Scholar]
- Lee, S.K.; Mbwambo, Z.H.; Chung, H.S.; Luyengi, L.; Gamez, E.J.C.; Mehta, R.G.; Kinghorn, A.D.; Pezzuto, J.M. Evaluation of the antioxidant potential of natural products. Comb. Chem. High Throughput Screen. 1998, 1, 35–46. [Google Scholar]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kondratyuk, T.P.; Park, E.-J.; Yu, R.; Van Breemen, R.B.; Asolkar, R.N.; Murphy, B.T.; Fenical, W.; Pezzuto, J.M. Novel Marine Phenazines as Potential Cancer Chemopreventive and Anti-Inflammatory Agents. Mar. Drugs 2012, 10, 451-464. https://doi.org/10.3390/md10020451
Kondratyuk TP, Park E-J, Yu R, Van Breemen RB, Asolkar RN, Murphy BT, Fenical W, Pezzuto JM. Novel Marine Phenazines as Potential Cancer Chemopreventive and Anti-Inflammatory Agents. Marine Drugs. 2012; 10(2):451-464. https://doi.org/10.3390/md10020451
Chicago/Turabian StyleKondratyuk, Tamara P., Eun-Jung Park, Rui Yu, Richard B. Van Breemen, Ratnakar N. Asolkar, Brian T. Murphy, William Fenical, and John M. Pezzuto. 2012. "Novel Marine Phenazines as Potential Cancer Chemopreventive and Anti-Inflammatory Agents" Marine Drugs 10, no. 2: 451-464. https://doi.org/10.3390/md10020451
APA StyleKondratyuk, T. P., Park, E. -J., Yu, R., Van Breemen, R. B., Asolkar, R. N., Murphy, B. T., Fenical, W., & Pezzuto, J. M. (2012). Novel Marine Phenazines as Potential Cancer Chemopreventive and Anti-Inflammatory Agents. Marine Drugs, 10(2), 451-464. https://doi.org/10.3390/md10020451