Shellfish Toxins Targeting Voltage-Gated Sodium Channels


Abstract
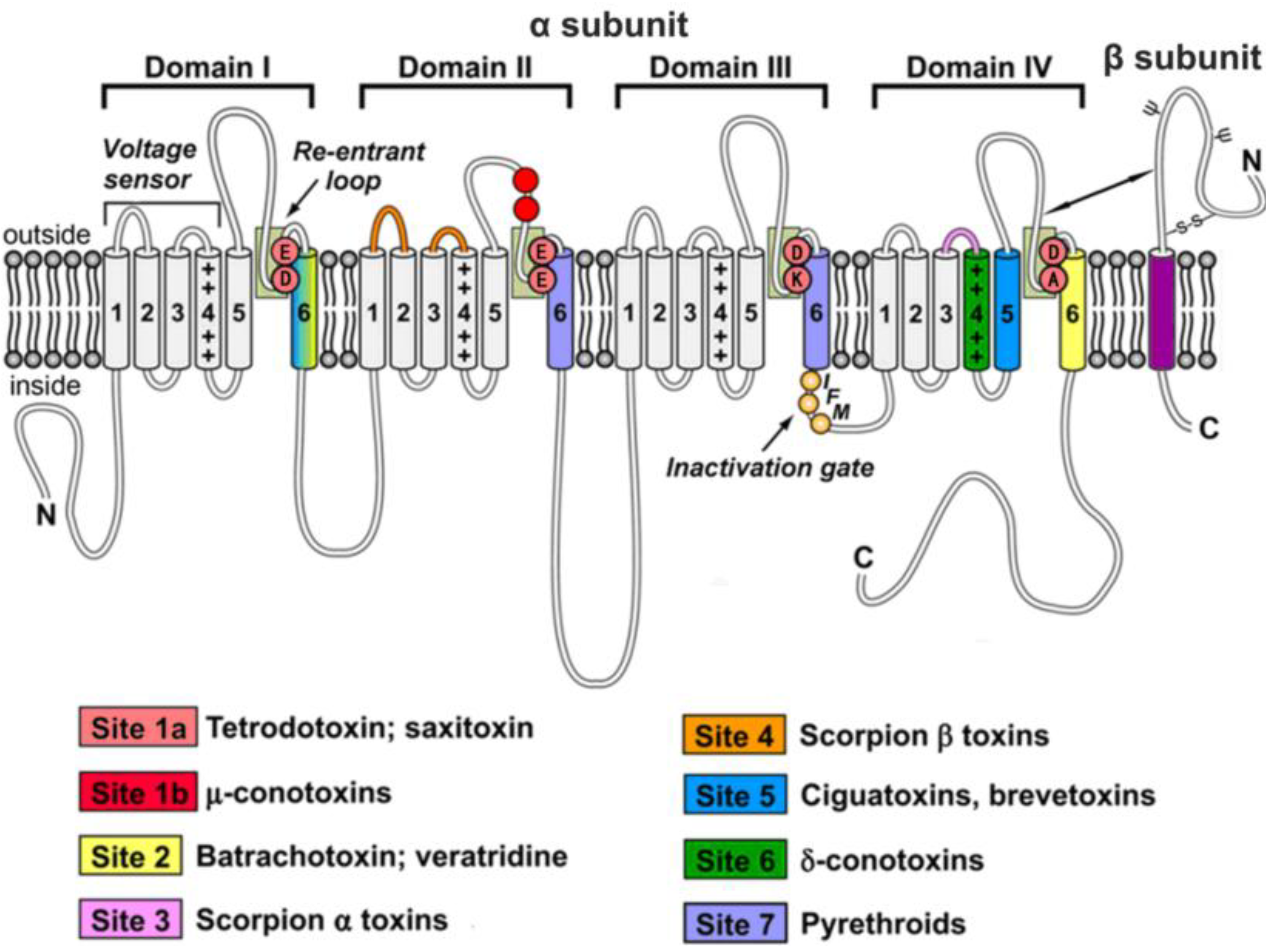
:1. Overview of Voltage-Gated Sodium Channels


| α-Subunits | Gene Symbol | Chromosomal Location1 ♦ | TTX-S/R ♦♦ | Predominant Location | Expression in DRG ♦♦♦ | Effect of Mutation |
|---|---|---|---|---|---|---|
| Nav1.1 | SCN1A | M:2 H:2q24 | S | CNS, PNS | +++ | Epilepsy |
| Nav1.2 | SCN2A | M:2 H:2q23-24 | S | CNS | + | Epilepsy |
| Nav1.3 | SCN3A | M:2 H:2q24 | S | CNS (embryonic) | upregulated after axotomy | None reported |
| Nav1.4 | SCN4A | M:11 H:17q23-25 | S | skeletal muscle | − | Myotonia, periodic paralysis |
| Nav1.5 | SCN5A | M:9 H:3p21 | R | heart muscle | − | Long-QT, Brugada syndrome, Progressive familial heart block |
| Nav1.6 | SCN8A | M:15 H:12q13 | S | CNS, PNS, glia nodes of Ranvier | +++ | Cerebellar atrophy |
| Nav1.7 | SCN9A | M:2 H:2q24 | S | PNS Schwann cell | +++ | Increased and decreased pain sensitivity |
| Nav1.8 | SCN10A | M:9 H:3p22-24 | R | PNS (sensory neurons) | +++ | None reported |
| Nav1.9 | SCN11A | M:9 H:3p21-24 | R | PNS | +++ | None reported |
| Nax | SCN6A (SCN7A) | M:2 H:2q21-23 | R | heart, uterus, glia, PNS smooth muscle | + | − |
2. Shellfish Toxins
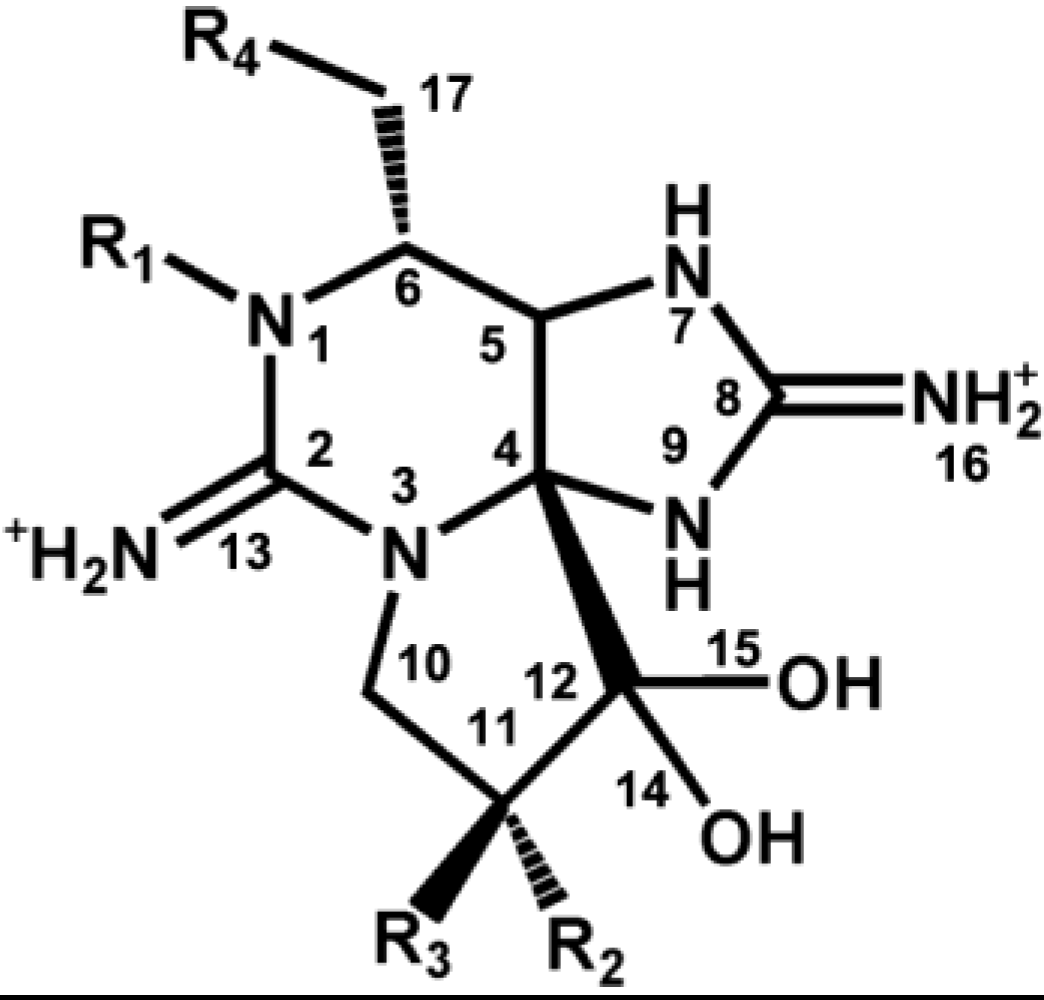
2.1. STX

| Toxin | R1 | R2 | R3 | R4 | |
|---|---|---|---|---|---|
| Carbamate | STX | H | H | H | OCONH2 |
| Neo STX | OH | H | H | OCONH2 | |
| GTX1 | OH | OSO3− | H | OCONH2 | |
| GTX2 | H | OSO3− | H | OCONH2 | |
| GTX3 | H | H | OSO3− | OCONH2 | |
| GTX4 | OH | H | OSO3− | OCONH2 | |
| N-sulfocarbamoyl | GTX5(B1) | H | H | H | OCONHSO3− |
| GTX6(B2) | OH | H | H | OCONHSO3− | |
| C1 | H | OSO3− | H | OCONHSO3− | |
| C2 | H | H | OSO3− | OCONHSO3− | |
| C3 | OH | OSO3− | H | OCONHSO3− | |
| C4 | OH | H | OSO3− | OCONHSO3− | |
| Decarbamoyl | dcSTX | H | H | H | OH |
| dcNeoSTX | OH | H | H | OH | |
| dcGTX1 | OH | OSO3− | H | OH | |
| dcGTX2 | H | OSO3− | H | OH | |
| dcGTX3 | H | H | OSO3− | OH | |
| dcGTX4 | OH | H | OSO3− | OH | |
| Deoxydecarbamoyl | doSTX | H | H | H | H |
| doGTX2 | H | H | OSO3− | H | |
| doGTX3 | H | OSO3− | H | H |
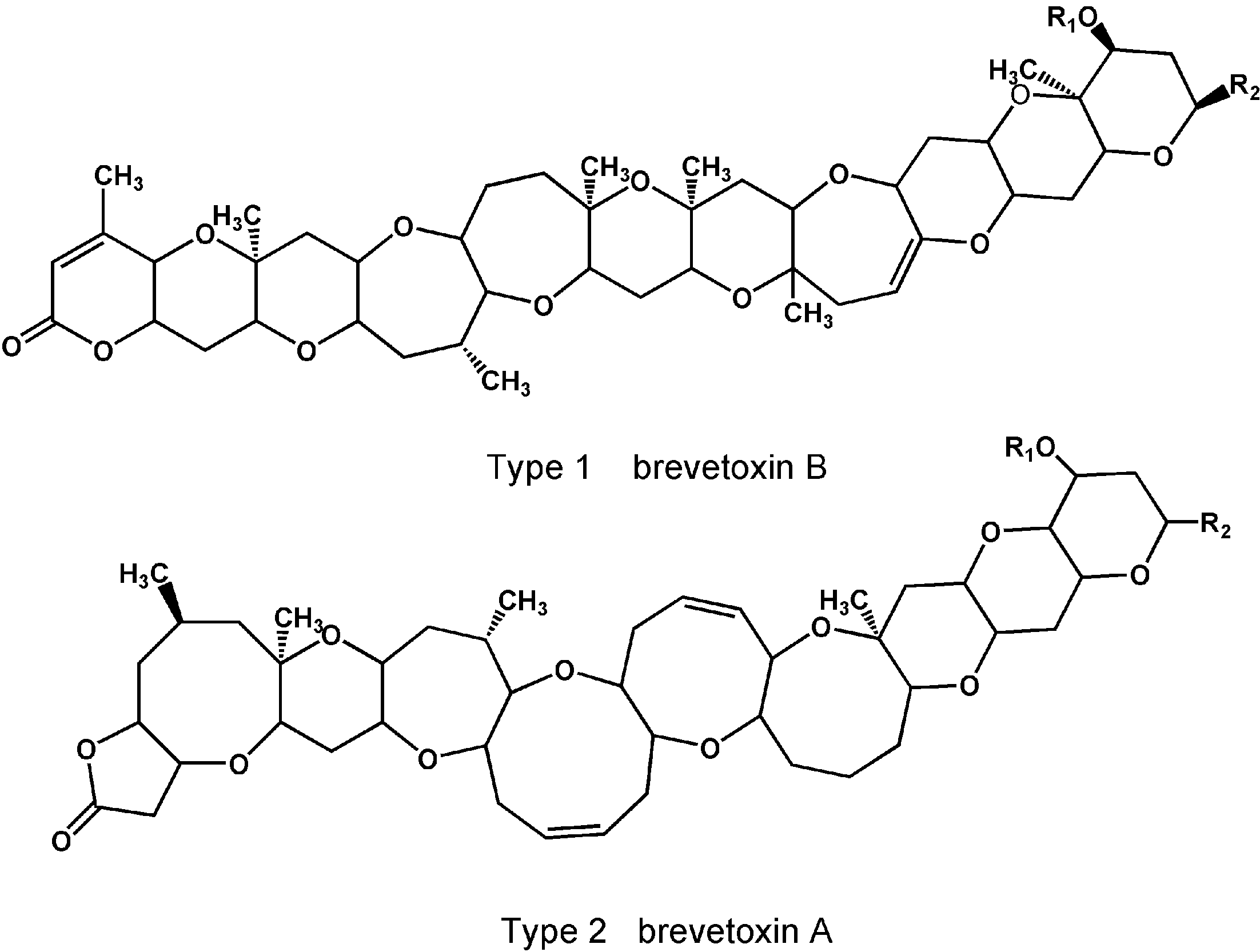
2.2. Brevetoxins

| Toxin | Type | R1 | R2 | Nominal Mass |
|---|---|---|---|---|
| PbTx-1 | 2 | H | CH2C(CH2)CHO | 866 |
| PbTx-2 | 1 | H | CH2C(CH2)CHO | 894 |
| PbTx-3 | 1 | H | CH2C(CH2)CH2OH | 896 |
| PbTx-5 | 1 | COCH3 | K-ring acetate PbTx-2 | 936 |
| PbTx-6 | 1 | H | K-ring epoxide PbTx-2 | 910 |
| PbTx-7 | 2 | H | CH2C(CH2)CH2OH | 868 |
| PbTx-8 | 1 | H | CH2COCH2Cl | 916 |
| PbTx-9 | 1 | H | CH2CH(CH3)CH2OH | 898 |
| PbTx-10 | 2 | H | CH2CH(CH3)CH2OH | 870 |
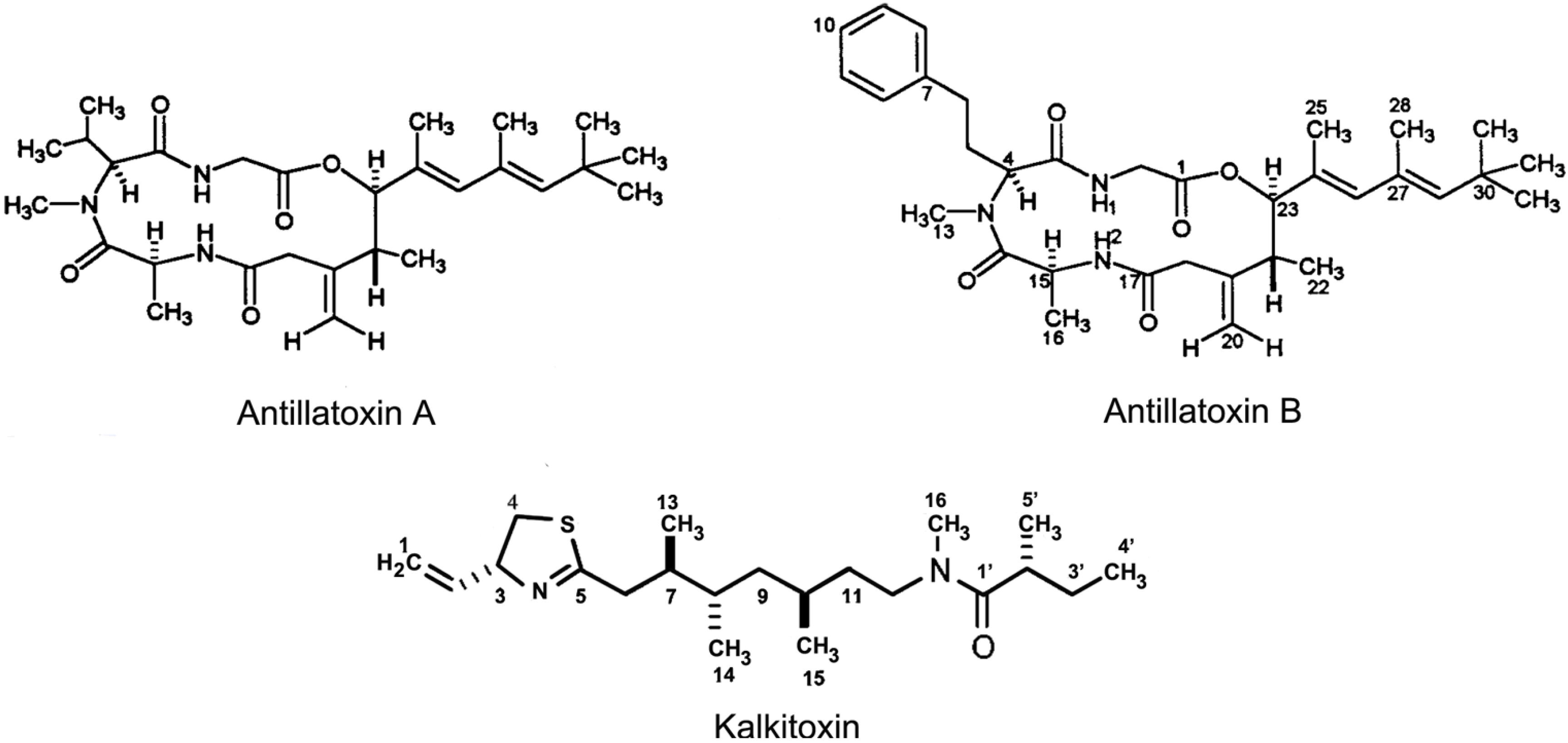
2.3. Antillatoxins

2.4. Kalkitoxin
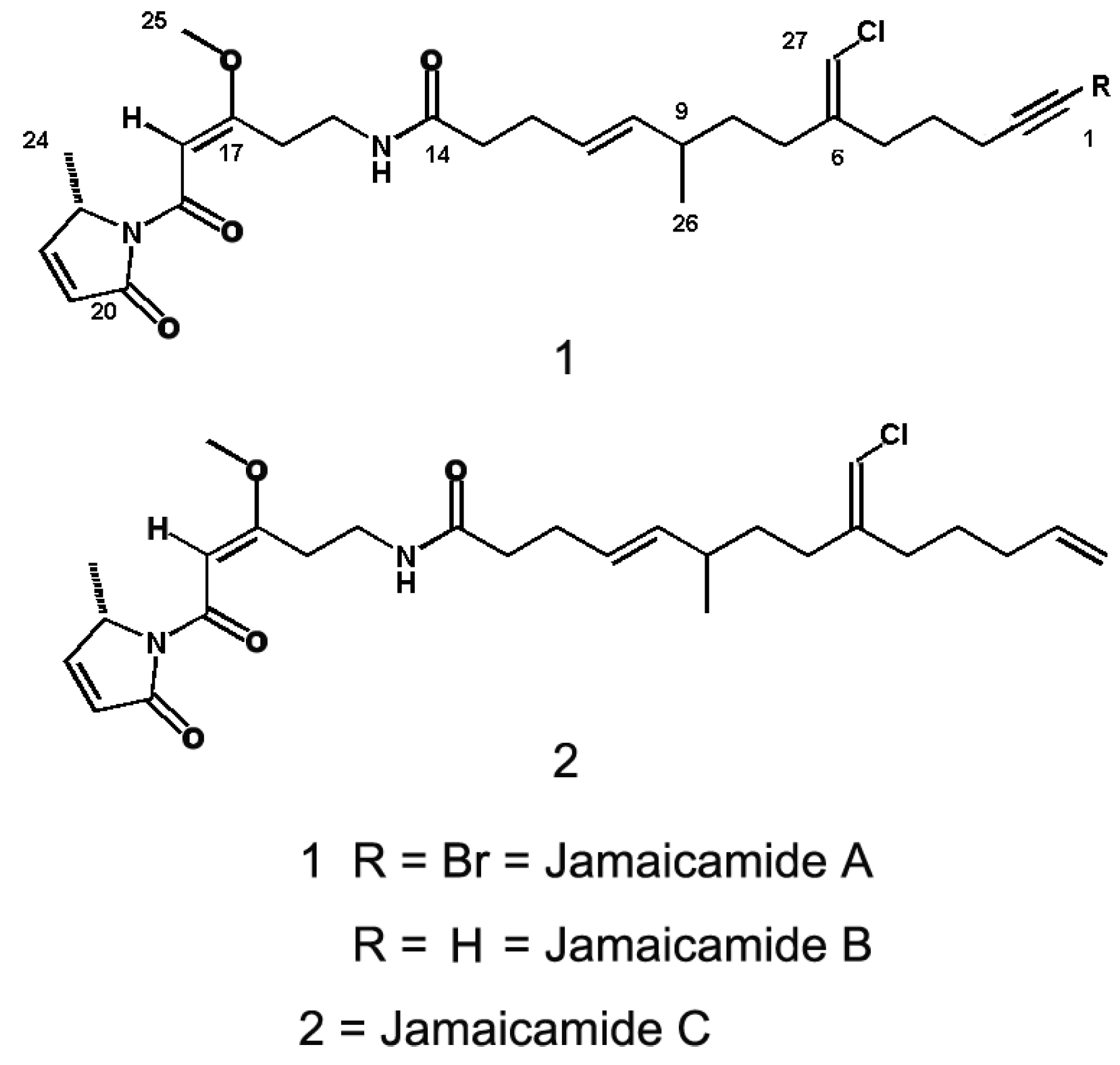
2.5. Jamaicamides

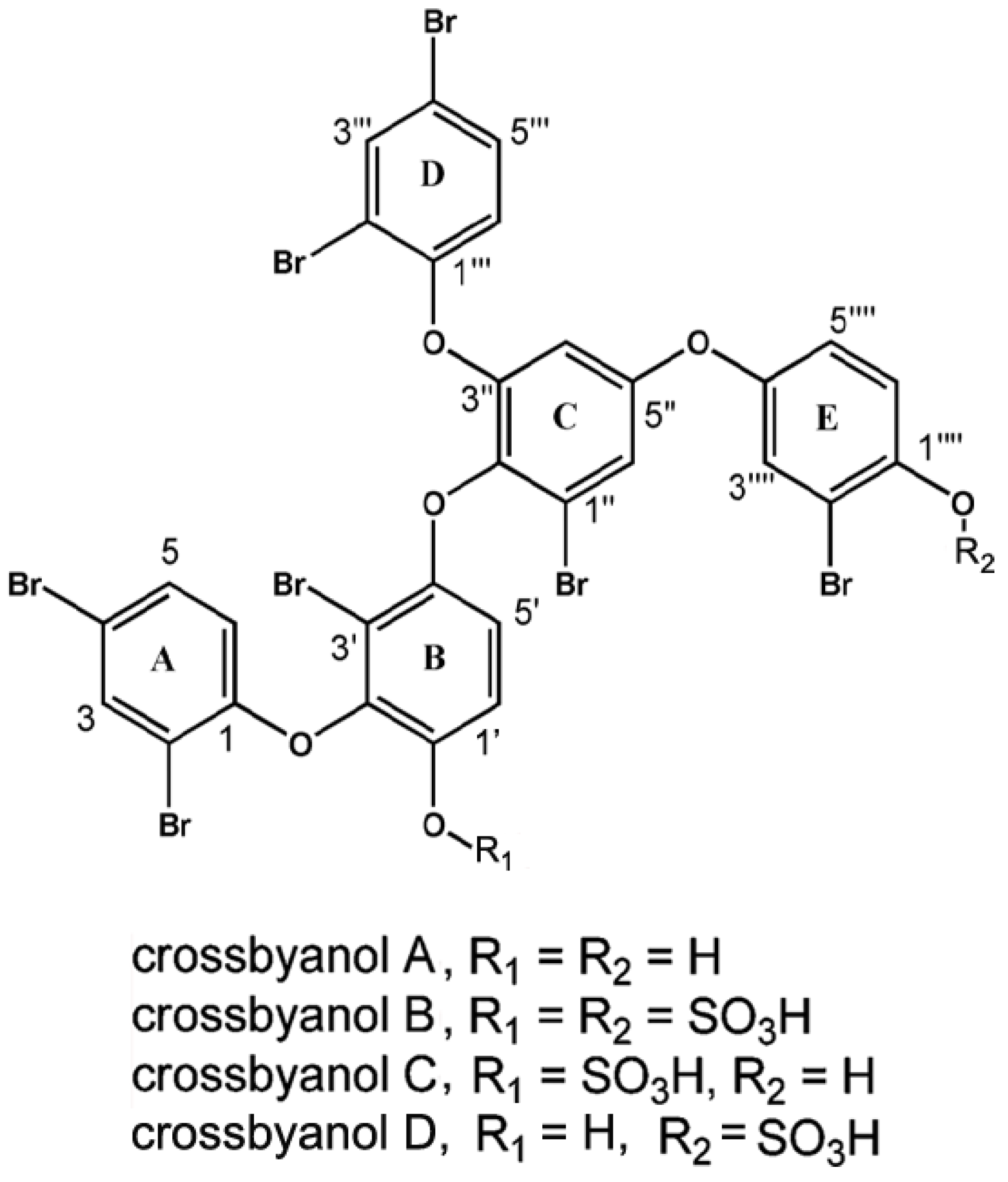
2.6. Crossbyanols

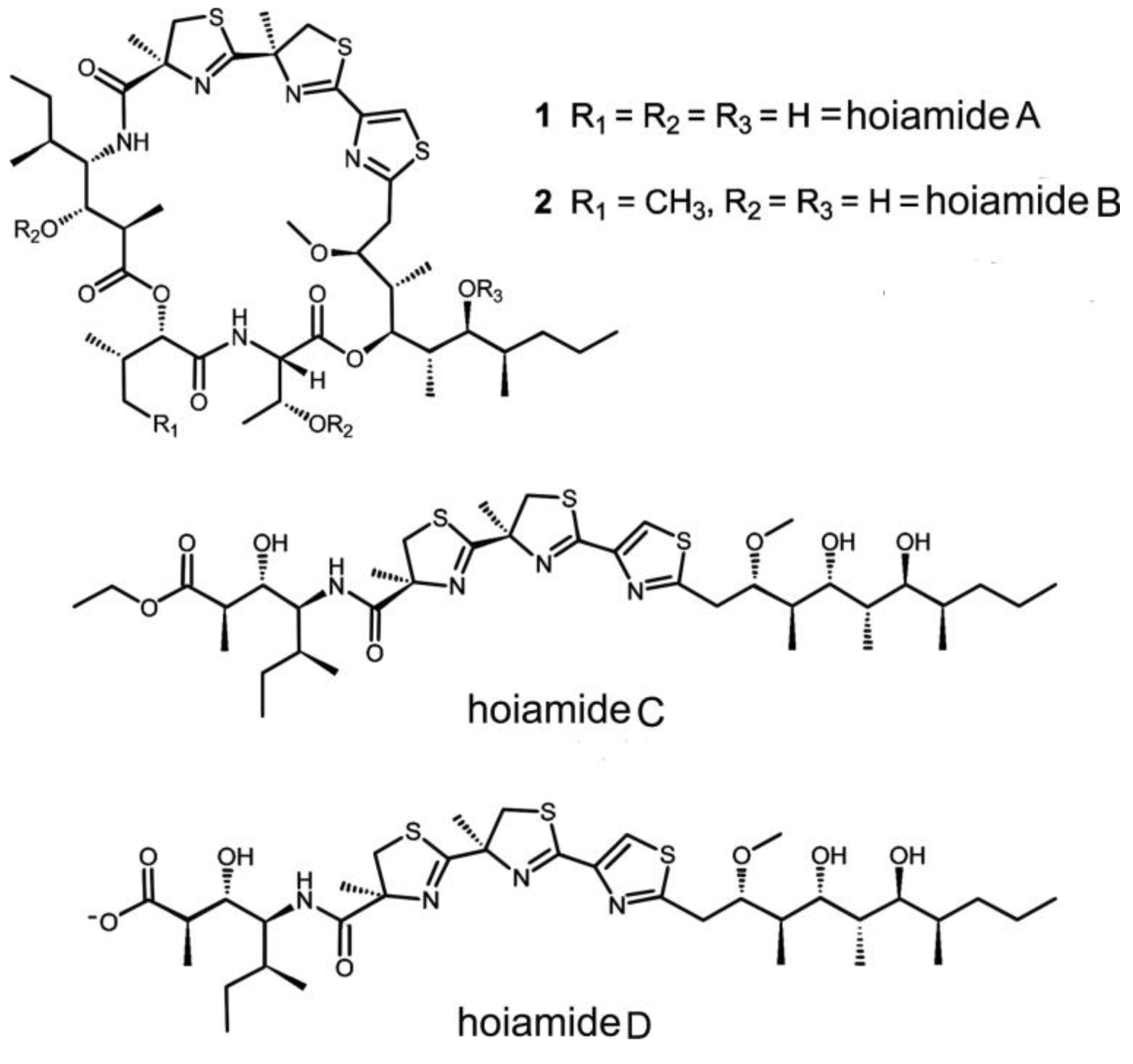
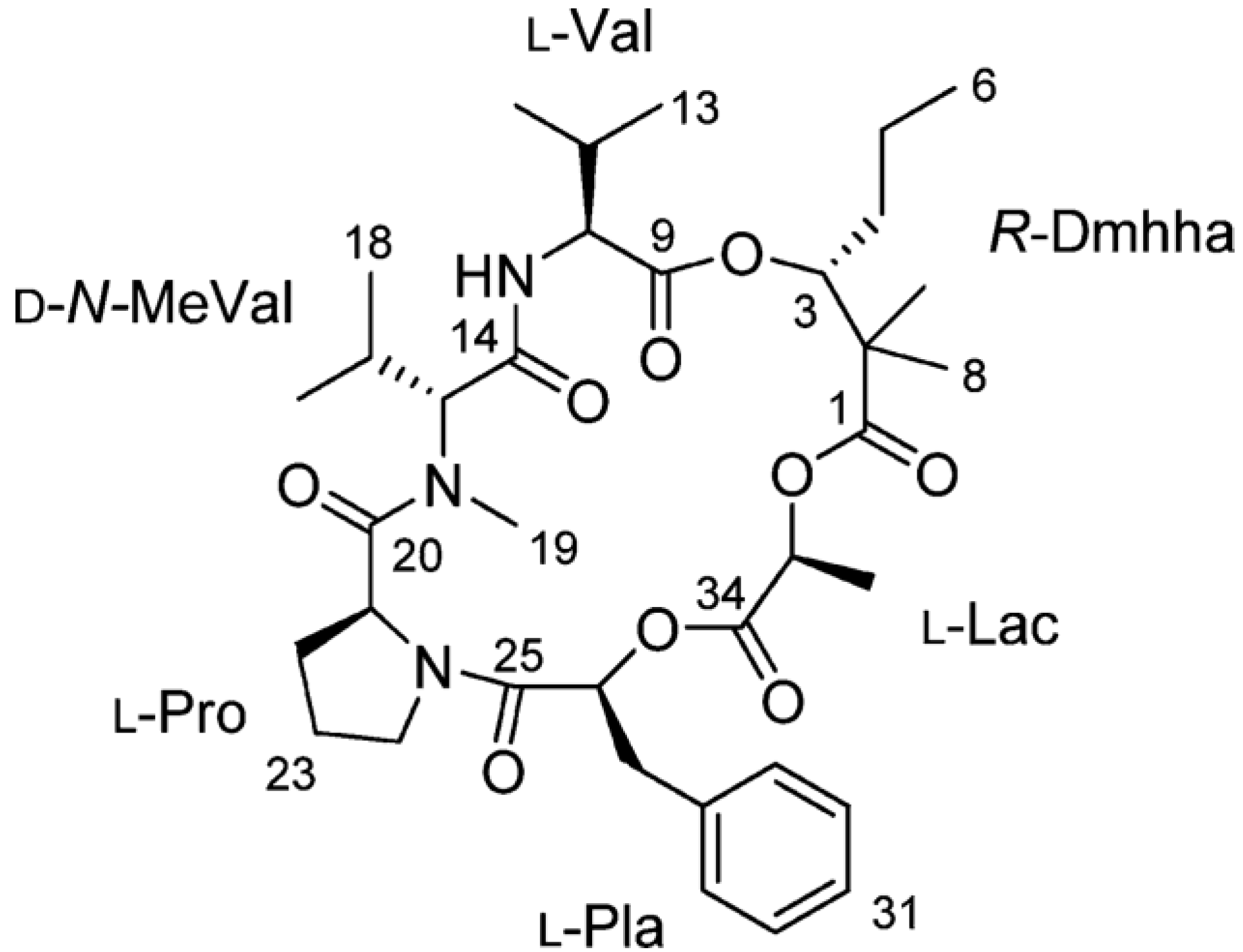
2.7. Hoiamides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
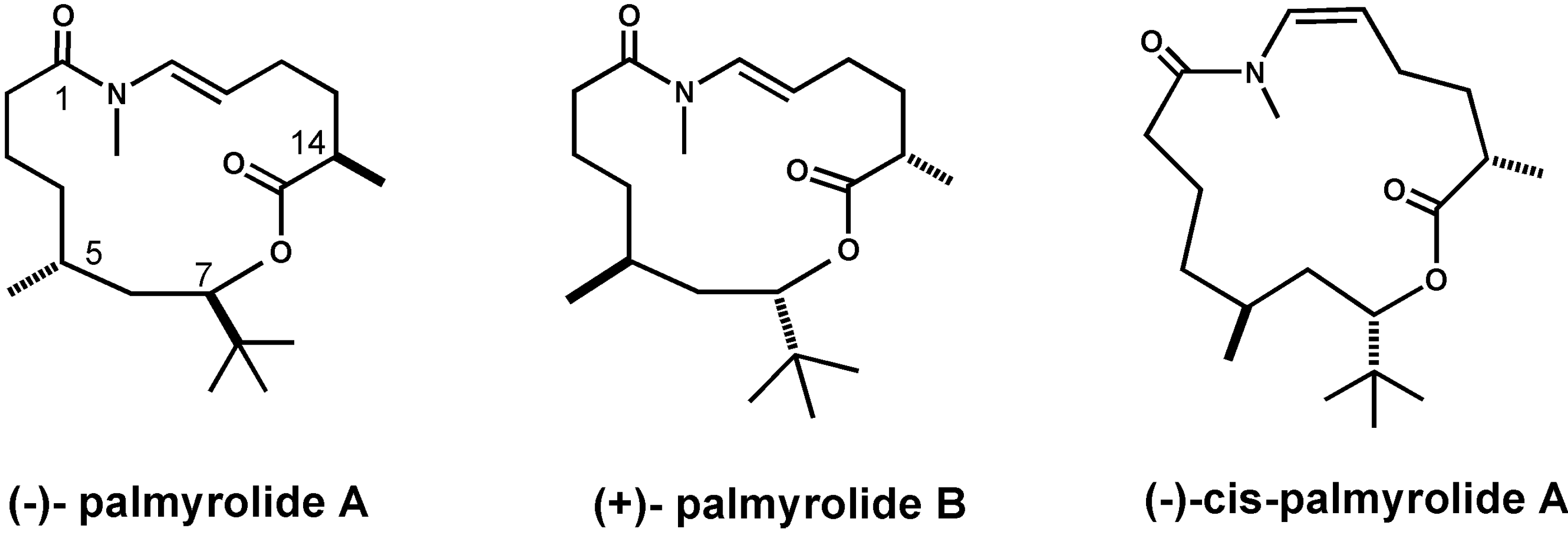
2.8. Palmyrolide A

2.9. Palmyramide A

3. Pharmaceutical Potential of Shellfish Toxins
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Cestèle, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar]
- Ogata, N.; Ohishi, Y. Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn. J. Pharmacol. 2002, 88, 365–377. [Google Scholar] [CrossRef]
- Isom, L.L.; DeJongh, K.S.; Patton, D.E.; Reber, B.F.; Offord, X.J.; Charbonneau, H.; Walsh, K.; Goldin, A.L.; Catterall, W.A. Primary structure and functional expression of the β1 Subunit of the rat brain sodium channel. Science 1992, 256, 839–842. [Google Scholar]
- Isom, L.L.; Ragsdale, D.S.; De Jongh, K.S.; Westenbroek, R.E.; Reber, B.F.; Scheuer, X.T.; Catterall, W.A. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 1995, 83, 433–442. [Google Scholar]
- Morgan, K.; Stevens, E.B.; Shah, B.; Cox, P.J.; Dixon, A.K.; Lee, K.; Pinnock, R.D.; Hughes, J.; Richardson, P.J.; Mizuguchi, K.; Jackson, A.P. β3: An additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc. Natl. Acad. Sci. USA 2000, 97, 2308–2313. [Google Scholar]
- Aman, T.K.; Grieco-Calub, T.M.; Chen, C.; Rusconi, R.; Slat, E.A.; Isom, L.L.; Raman, I.M. Regulation of persistent Na current by interactions between β subunits of voltage-gated Na channels. J. Neurosci. 2009, 29, 2027–2042. [Google Scholar]
- Patino, G.A.; Isom, L.L. Electrophysiology and beyond: multiple roles of Na+ channel β subunits in development and disease. Neurosci. Lett. 2010, 486, 53–59. [Google Scholar]
- Wang, J.; Yarov-Yarovoy, V.; Kahn, R.; Gordon, D.; Gurevitz, M.; Scheuer, M.; Catterall, W.A. Mapping the receptor site for α-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. USA 2011, 108, 15426–15431. [Google Scholar]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [Green Version]
- King, G.F.; Escoubas, P.; Nicholson, G.M. Peptide toxins that selectively target insect NaV and CaV channels. Channels 2008, 2, 100–116. [Google Scholar]
- Catterall, W.A. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 2010, 67, 915–928. [Google Scholar] [CrossRef]
- Smith, M.R.; Goldin, A.L. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophys. J. 1997, 73, 1885–1895. [Google Scholar] [CrossRef]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Brackenbury, W.J.; Isom, L.L. Voltage-gated Na+ channels: Potential for β subunits as therapeutic targets. Expert Opin. Ther. Targets 2008, 12, 1191–1203. [Google Scholar] [CrossRef]
- Mantegazza, M.; Curia, G.; Biagini, G.; Ragsdale, D.S.; Avoli, M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010, 9, 413–424. [Google Scholar]
- England, S.; de Groot, M.J. Subtype-selective targeting of voltage-gated sodium channels. Br. J. Pharmacol. 2009, 158, 1413–1425. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Ogta, N.; Tatebayashi, H. Kinetic analysis of two types of Na+ channels in rat dorsal root ganglia. J. Physiol. 1993, 466, 9–37. [Google Scholar]
- Goldin, A.L. Resurgence of sodium research. Annu. Rev. Physiol. 2001, 63, 871–894. [Google Scholar] [CrossRef]
- Fainzilber, M.; Kofman, O.; Zlotkin, E.; Gordon, D. A new neurotoxin receptor site on sodium channels is identified by a conotoxin that affects sodium channel inactivation in molluscs and acts as an antagonist in rat brain. J. Biol. Chem. 1994, 269, 2574–2680. [Google Scholar]
- Lefebvre, K.A.; Bill, B.D.; Erickson, A.; Baugh, K.A.; O’Rourke, L.; Costa, P.R.; Nance, S.; Trainer, V.L. Characterization of intracellular and extracellular saxitoxin levels in both field and cultured Alexandrium spp. samples from Sequim Bay, Washington. Mar. Drugs 2008, 6, 103–116. [Google Scholar]
- Usup, G.; Kulis, D.M.; Anderson, D.M. Growth and toxin production of the toxic dinoflasellate Pyodinium bahamense var. compressum in laboratory cultures. Nat. Toxins 1994, 2, 254–262. [Google Scholar]
- Oshima, Y.; Blackburn, S.I.; Hallegraeff, G.M. Comparative study on paralytic shellfish toxin of the dinoflagellate Gymnodinium catenatum from three different countries profiles. Mar. Biol. 1993, 116, 471–476. [Google Scholar]
- Daugbjerg, N.; Hansen, G.; Larsen, J.; Mosestrup, Ø. Phylogeny of some of the major genera of dinoflagellates based on ultrastructure and partial LSU rDNA sequence data, including the erection of three new genera of unarmoured dinoflagellates. Phycologia 2000, 39, 302–317. [Google Scholar] [CrossRef]
- Smith, F.M.; Wood, S.A.; Ginkel, R.V.; Broady, P.A.; Gaw, S. First report of saxitoxin production by a species of the freshwater benthic cyanobacterium, Scytonema agardh. Toxicon 2011, 57, 566–573. [Google Scholar]
- Wang, D.Z. Neurotoxins from marine dinoflagellates: A brief review. Mar. Drugs 2008, 6, 349–371. [Google Scholar] [CrossRef]
- Cusick, K.D.; Sayler, G.S. An overview on the marine neurotoxin, saxitoxin: Genetics, molecular targets, methods of detection and ecological functions. Mar. Drugs 2013, 11, 991–1018. [Google Scholar]
- Watkins, S.M.; Reich, A.; Fleming, L.E.; Hammond, R. Neurotoxic shellfish poisoning. Mar. Drugs 2008, 6, 431–455. [Google Scholar] [CrossRef]
- Van Dolah, F.M. Marine algal toxins: Origins, health effects, and their increased occurrence. Environ. Health Perspect. 2000, 108, 133–141. [Google Scholar]
- Anderson, D.M.; Glibert, P.M.; Burkholder, J.M. Harmful algal blooms and eutrophication: Nutrient sources, composition, and consequences. Estuaries 2002, 25, 704–726. [Google Scholar]
- Sellner, K.G.; Doucette, G.J.; Kirkpatrick, G.J. Harmful algal blooms: causes, impacts and detection. J. Ind. Microbiol. Biotechnol. 2003, 30, 383–406. [Google Scholar] [CrossRef]
- Rogart, R.B. High-STX-affinity vs. low-STX-affinity Na+ channel subtypes in nerve, heart, and skeletal muscle. Ann. N. Y. Acad. Sci. 1986, 479, 402–430. [Google Scholar]
- Wang, J.; Salata, J.J.; Bennett, P.B. Saxitoxin is a gating modifier of hERG K+ channels. J. Gen. Physiol. 2003, 121, 583–598. [Google Scholar]
- Su, Z.; Sheets, M.; Ishida, H.; Li, F.; Barry, W.H. Saxitoxin blocks L-type ICa. J. Pharmacol. Exp. Ther. 2004, 308, 324–329. [Google Scholar]
- Böttinger, H.; Béress, L.; Habermann, E. Involvement of (Na+ + K+)-ATPase in binding and actions of palytoxin on human erythrocytes. Biochim. Biophys. Acta Biomembr. 1986, 861, 165–176. [Google Scholar]
- Thomas, P.; Stephens, M.; Wilkie, G.; Amar, M.; Lunt, G.G.; Whiting, P.; Gallagher, T.; Pereira, E.; Alkondon, M.; Albuquerque, E.X.; Wonnacott, S. (+)-Anatoxin-a is a potent agonist at neuronal nicotinic acetylcholine receptors. J. Neurochem. 1993, 60, 2308–2311. [Google Scholar]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar]
- Schantz, E.J.; Mold, J.D.; Stanger, D.W.; Shavel, J.; Riel, F.J.; Bowden, J.P.; Lynch, J.M.; Wyler, R.S.; Riecel, B.; Sommer, H. Paralytic shellfish poison. VI. A procedure for the isolation and purification of the poison from toxic clam and mussel tissues. J. Am. Chem. Soc. 1957, 79, 5230–5235. [Google Scholar] [CrossRef]
- Terlau, H.; Heinemann, S.H.; Stühmer, W.; Pusch, M.; Conti, F.; Imoto, K.; Numa, S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991, 293, 93–96. [Google Scholar] [CrossRef]
- Noda, M.; Suzuki, H.; Numa, S.; Stiihmer, W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989, 259, 213–216. [Google Scholar]
- Barnet, C.S.; Tse, J.Y.; Kohane, D.S. Site 1 sodium channel blockers prolong the duration of sciatic nerve blockade from tricyclic antidepressants. Pain 2004, 110, 432–438. [Google Scholar] [CrossRef]
- Garrido, R.; Lagos, N.; Lagos, M.; Rodríguez-Navarro, A.J.; Garcia, C.; Truan, D.; Henriquez, A. Treatment of chronic anal fissure by gonyautoxin. Colorectal Dis. 2007, 9, 619–624. [Google Scholar]
- Lattes, K.; Venegas, P.; Lagos, N.; Lagos, M.; Pedraza, L.; Rodriguez-Navarro, A.J.; García, C. Local infiltration of gonyautoxin is safe and effective in treatment of chronic tension-type headache. Neurol. Res. 2009, 31, 228–233. [Google Scholar] [CrossRef]
- Negri, A.P.; Jones, G.J.; Hindmarsh, M. Sheep mortality associated with paralytic shellfish poisons from the cyanbacterium Anabaena circinalis. Toxicon 1995, 33, 1321–1329. [Google Scholar]
- Reyero, M.; Cacho, E.; Martínez, A.; Vázquez, J.; Marina, A,; Fraga, S.; Franco, J.M. Evidence of saxitoxin derivatives as causative agents in the 1997 mass mortality of monk seals in the Cape Blanc Peninsula. Nat. Toxins 1999, 7, 311–315. [Google Scholar]
- Anderson, D.M. Red tides. Sci. Am. 1994, 271, 62–68. [Google Scholar] [CrossRef]
- Stewart, I.; Seawright, A.A.; Shaw, G.R. Cyanobacterial poisoning in livestock, wild mammals and birds—an overview. In Cyanobacterial Harmful Algal Blooms: State of the Science and Research Needs; Springer Science and Business Media LLC: New York, NY, USA, 2008; pp. 613–637. [Google Scholar]
- Trainer, V.L.; Baden, D.G. High affinity binding of red tide neurotoxins to marine mammal brain. Aquat. Toxicol. 1999, 46, 139–148. [Google Scholar]
- Halsetead, B.W. Poisonous and Venomous Marine Animals of the World; Darwin Press: Princeton, NJ, USA, 1978; pp. 325–400. [Google Scholar]
- Llewellyn, L.E. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 2006, 23, 200–222. [Google Scholar] [CrossRef]
- Pearson, L.; Mihali, T.; Moffitt, M.; Kellmann, R.; Neilan, B. On the chemistry, toxicology and genetics of the cyanobacterial toxins, microcystin, nodularin, saxitoxin and cylindrospermopsin. Mar. Drugs 2010, 8, 1650–1680. [Google Scholar]
- Lagos, N.; Andrinolo, D. Paralytic shellfish poisoning (PSP): Toxicology and kinetics. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; Botana, L.M., Ed.; Marcel Dekker: New York, NY, USA, 2000; pp. 203–215. [Google Scholar]
- Kao, C.Y. Paralytic shellfish poisoning. In Algal Toxins in Seafood and Drinking Water; Falconer, I.R., Ed.; Academic Press: London, UK, 1993; pp. 75–86. [Google Scholar]
- Hall, S.; Strichartz, G.; Moczydlowski, E.; Ravindran, A.; Reichardt, P.B. The saxitoxins: Sources, chemistry and pharmacology. In Marine Toxins. Origin, Structure and Pharmacology; Hall, S., Reichardt, P.B., Eds.; American Chemical Society: Washington, DC, USA, 1990; pp. 29–69. [Google Scholar]
- Schantz, E.J.; Ghazarossian, V.E.; Schnoes, H.K.; Strong, F.M.; Springer, J.P.; Pezzanite, J.O.; Clardy, J. The structure of saxitoxin 1. J. Am. Chem. Soc. 1975, 97, 1238–1239. [Google Scholar]
- Bordner, J.; Thiessen, W.E.; Bates, H.A.; Rapoport, H. The structure of a crystalline derivative of saxitoxin. The structure of saxitoxin. J. Am. Chem. Soc. 1975, 97, 6008–6012. [Google Scholar]
- Rogers, S.R.; Rapport, H. The pKa’s of saxitoxin. J. Am. Chem. Soc. 1980, 102, 7335–7339. [Google Scholar] [CrossRef]
- Shimizu, Y.; Hsu, C.P.; Genenah, A. Structure of saxitoxin in solutions and stereochemistry of dihydrosaxitoxins. J. Am. Chem. Soc. 1981, 103, 605–609. [Google Scholar] [CrossRef]
- Schantz, E.J.; Lynch, J.M.; Vayvada, G.; Matsumoto, K.; Rapoport, H. The purification and characterization of the poison produced by Gonyaulax catenella in axenic culture. Biochemistry 1966, 5, 1191–1195. [Google Scholar]
- Strichartz, G. Structural determinants of the affinity of saxitoxin for neuronal sodium Channels. Electrophysiological studies on frog peripheral nerve. J. Gen. Physiol. 1984, 84, 281–305. [Google Scholar] [CrossRef]
- Van Dolah, F.M. Diversity of Marine and Freshwater Algal Toxins. In Seafood Toxicology: Pharmacology, Physiology and Detection; Botana, L., Ed.; Marcel Dekker: New York, NY, USA, 2000; pp. 19–43. [Google Scholar]
- Hille, B. The receptor for tetrodotoxin and saxitoxin. A structural hypothesis. Biophys. J. 1975, 15, 615–619. [Google Scholar] [CrossRef]
- Kao, C.Y.; Walker, S.E. Active groups of saxitoxin and tetrodotoxin as deduced from actions of saxitoxin analogues on frog muscle and squid axon. J. Physiol. 1982, 323, 619–637. [Google Scholar]
- Shiinizu, Y. Recent progress in marine toxin research. Pure Appl. Chem. 1982, 54, 1973–1980. [Google Scholar]
- Numa, S.; Noda, M. Molecular structure of sodium channels. Ann. N. Y. Acad. Sci. 1986, 479, 338–355. [Google Scholar] [CrossRef]
- Hartshorne, R.P.; Catterall, W.A. The sodium channel from rat brain. Purification and subunit composition. J. Biol. Chem. 1984, 259, 1667–1675. [Google Scholar]
- Walker, J.R.; Novick, P.A.; Parsons, W.H.; McGregor, M.; Zablocki, J.; Pande, V.S.; Du Bois, J. Marked difference in saxitoxin and tetrodotoxin affinity for the human nociceptive voltage-gated sodium channel (Nav1.7). Proc. Natl. Acad. Sci. USA 2012, 109, 18102–18107. [Google Scholar]
- Steidinger, K.A. Phytoplankton ecology: A conceptual review based on eastern. Gulf of Mexico research. Crit. Rev. Microbiol. 1973, 3, 49–68. [Google Scholar] [CrossRef]
- Baden, D.G. Marine food-borne dinoflagellate toxins. Int. Rev. Cytol. 1983, 82, 99–150. [Google Scholar]
- Baden, D.G.; Mende, T.J. Toxicity of two toxins from the Florida red tide marine dinoflagellate, Ptychodiscus brevis. Toxicon 1982, 20, 457–461. [Google Scholar] [CrossRef]
- Poli, M.; Mende, T.J.; Baden, D.G. Brevetoxins, unique activators of voltage-sensitive sodium channels bind to specific sites in rat brain synaptosomes. Mol. Pharmacol. 1986, 30, 129–135. [Google Scholar]
- Baden, D.; Fleming, L.E.; Bean, J.A. Marine Toxins. In Handbook of Clinical Neurology: Intoxications of the Nervous System Part H. Natural Toxins and Drugs; deWolf, F.A, Ed.; Elsevier: Amsterdam, The Nertherlands, 1995; pp. 141–175. [Google Scholar]
- Landsberg, J.H. The effects of harmful algal blooms on aquatic organisms. Rev. Fish. Sci. 2002, 10, 113–390. [Google Scholar]
- Flewelling, L.J.; Naar, J.P.; Abbott, J.P.; Baden, D.G.; Barros, N.B.; Bossart, G.D.; Bottein, M.Y.; Hammond, D.G.; Haubold, E.M.; Heil, C.A.; et al. Brevetoxicosis: Red tides and marine mammal mortalities. Nature 2005, 435, 755–756. [Google Scholar]
- Sagir Ahmed, M.D.; Arakawa, O.; Onoue, Y. Toxicity of cultured Chatonella marina. In Harmful Marine Algal Blooms; Lassus, P., Arzul, G., Erhard, E., Gentien, P., Marcaillou, C., Eds.; Lavoisier: Paris, France, 1995; pp. 499–504. [Google Scholar]
- Khan, S.; Arakawa, O.; Onoue, Y. Neurotoxins in a toxic red tide of Heterosigma akashiwo (Raphidophyceae) in Kagoshima Bay, Japan. Aquac. Res. 1997, 28, 9–14. [Google Scholar] [CrossRef]
- Hallegraeff, G.M.; Munday, B.L.; Baden, D.G.; Whitney, P.L. Chatonnella maria raphidophyte bloom associated with mortality of cultured bluefin tuna (Thunnus maccoyii) in south Australia. In Harmful Algae; Reguera, B., Blanco, J., Ferandz, M.L., Wyatt, T., Eds.; Xunta de Galacia and IOC: Santiago de Compostela, Spain, 1998; pp. 93–96. [Google Scholar]
- Morris, P.D.; Campbell, D.S.; Taylor, T.J.; Freeman, J.I. Clinical and epidemiological features of neurotoxic shellfish poisoning in North Carolina. Am. J. Public Health 1991, 81, 471–474. [Google Scholar]
- Kirkpatrick, B.; Fleming, L.E.; Squicciarini, D.; Backer, L.C.; Clark, R.; Abraham, W.; Benson, J.; Chenge, Y.S.; Johnson, D.; Pierce, R.; et al. Literature review of Florida red tide: Implications for human health effects. Harmful Algae 2004, 3, 99–115. [Google Scholar] [CrossRef]
- Gallagher, J.P.; Shinnick-Gallaqher, P. Effect of Gymnodinium breve toxin in the rat phrenic nerve diaphragm preparation. Br. J. Pharmacol. 1980, 69, 367–372. [Google Scholar]
- Trainer, V.L.; Thomsen, W.J.; Catterall, W.A.; Baden, D.G. Photoaffinity labeling of the brevetoxin receptor on sodium channels in rat brain synaptosomes. Mol. Pharmacol. 1991, 40, 988–994. [Google Scholar]
- Rein, K.S.; Baden, D.G.; Gawley, R.E. Conformational analysis of the sodium channel modulator, brevetoxin A, comparison with brevetoxin B conformations, and a hypothesis about the common pharmacophore of the “site 5” toxins. J. Org. Chem. 1994, 59, 2101–2106. [Google Scholar] [CrossRef]
- Baden, D.G.; Bourdelais, A.J.; Jacocks, H.; Michelliza, S.; Naar, J. Natural and derivative brevetoxins: Historical background, multiplicity, and effects. Environ. Health Perspect. 2005, 113, 621–625. [Google Scholar]
- Orjala, J.; Nagle, D.G.; Hsu, V.L.; Gerwick, W.H. Antillatoxin: An exceptionally ichthyotoxic cyclic lipopeptide from the tropical cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 1995, 117, 8281–8282. [Google Scholar]
- Yokokawa, F.; Fujiwara, H.; Shioiri, T. Total synthesis and revision of absolute stereochemistry of antillatoxin, an ichthyotoxic cyclic lipopeptide from marine cyanobacterium Lyngbya majuscula. Tetrahedron 2000, 56, 1759–1775. [Google Scholar] [CrossRef]
- Li, W.I.; Marquez, B.L.; Okino, T.; Yokokawa, F.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. Characterization of the preferred stereochemistry for the neuropharmacologic actions of antillatoxin. J. Nat. Prod. 2004, 67, 559–568. [Google Scholar]
- Osborne, N.J.; Webb, P.M.; Shaw, G.R. The toxins of Lyngbya majuscula and their human and ecological health effects. Environ. Int. 2001, 27, 381–392. [Google Scholar] [CrossRef]
- Nogle, L.M.; Okino, T.; Gerwick, W.H. Antillatoxin B, a neurotoxic lipopeptide from the marine cyanobacterium lyngbya majuscule. J. Nat. Prod. 2001, 64, 983–985. [Google Scholar] [CrossRef]
- Wu, M.; Okino, T.; Nogle, L.M.; Marquez, B.L.; Williamson, R.T.; Sitachitta, N.; Berman, F.W.; Murray, T.F.; McGough, K.; Jacobs, R.; et al. Structure, synthesis, and biological properties of kalkitoxin, a novel neurotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2000, 122, 12041–12042. [Google Scholar]
- Aráoz, R.; Molgó, J.; Tandeau de Marsac, N. Neurotoxic cyanobacterial toxins. Toxicon 2010, 56, 813–828. [Google Scholar] [CrossRef]
- Yokokawa, F.; Fujiwara, H.; Shioiri, T. Total synthesis and revision of absolute configuration of antillatoxin, an ichthyotoxic cyclic lipopeptide of marine origin. Tetrahedron Lett. 1999, 40, 1915–1916. [Google Scholar] [CrossRef]
- Yokokawa, F.; Shioiri, T. Total synthesis of antillatoxin, an ichthyotoxic cyclic lipopeptide, having the proposed structure. What is the real structure of antillatoxin? J. Org. Chem. 1998, 63, 8638–8639. [Google Scholar] [CrossRef]
- Berman, F.W.; Gerwick, W.H.; Murray, T.F. Antillatoxin and kalkitoxin, ichthyotoxins from the tropical cyanobacterium Lyngbya majuscula, induce distinct temporal patterns of NMDA receptor-mediated neurotoxicity. Toxicon 1999, 37, 1645–1648. [Google Scholar] [CrossRef]
- Li, W.I.; Berman, F.W.; Okino, T.; Yokokawa, F.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. Antillatoxin is a marine cyanobacterial toxin that potently activates voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA 2001, 98, 7599–7604. [Google Scholar]
- Cao, Z.; George, J.; Gerwick, W.H.; Baden, D.G.; Rainier, J.D.; Murray, T.F. Influence of lipid-soluble gating modifier toxins on sodium influx in neocortical neurons. J. Pharmacol. Exp. Ther. 2008, 326, 604–613. [Google Scholar]
- Jabba, S.V.; Prakash, A.; Dravid, S.M.; Gerwick, W.H.; Murray, T.F. Antillatoxin, a novel lipopeptide, enhances eeurite outgrowth in immature cerebrocortical neurons through activation of voltage-gated sodium channels. J. Pharmacol. Exp. Ther. 2010, 332, 698–709. [Google Scholar]
- Cao, Z.; Gerwick, W.H.; Murray, T.F. Antillatoxin is a sodium channel activator that displays unique efficacy in heterologously expressed rNav1.2, rNav1.4 and rNav1.5 alpha subunits. BMC Neurosci. 2010, 11, 1–13. [Google Scholar]
- Nogle, L.M.; Gerwick, W.H. Diverse secondary metabolites from a Puerto Rican collection of Lyngbya majuscula. J. Nat. Prod. 2003, 66, 217–220. [Google Scholar] [CrossRef]
- White, J.D.; Lee, C.S.; Xu, Q. Total synthesis of (+)-kalkitoxin. Chem. Commun. (Camb.) 2003, 16, 2012–2013. [Google Scholar]
- White, J.D.; Xu, Q.; Lee, C.S.; Valeriote, F.A. Total synthesis and biological evaluation of (+)-kalkitoxin, a cytotoxic metabolite of the cyanobacterium Lyngbya majuscule. Org. Biomol. Chem. 2004, 2, 2092–2102. [Google Scholar]
- Umezawa, T.; Sueda, M.; Kamura, T.; Kawahara, T.; Han, X.; Okino, T.; Matsuda, F. Synthesis and biological activity of kalkitoxin and its analogues. J. Org. Chem. 2012, 77, 357–370. [Google Scholar]
- LePage, K.T.; Goeger, D.; Yokokawa, F.; Asano, T.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. The neurotoxic lipopeptide kalkitoxin interacts with voltage-sensitive sodium channels in cerebellar granule neurons. Toxicol. Lett. 2005, 158, 133–139. [Google Scholar]
- Edwards, D.J.; Marquez, B.L.; Nogle, L.M.; McPhail, K.; Goeger, D.E.; Roberts, M.A.; Gerwick, W.H. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscule. Chem. Biol. 2004, 11, 817–833. [Google Scholar]
- Graf, K.M.; Tabor, M.G.; Brown, M.L.; Paige, M. Synthesis of (s)-jamaicamide C carboxylic acid. Org. Lett. 2009, 11, 5382–5385. [Google Scholar] [CrossRef]
- Manger, R.L.; Leja, L.S.; Lee, S.Y.; Hungerford, J.M.; Hokama, Y.; Dickey, R.W.; Granade, H.R.; Lewis, R.; Yasumoto, T.; Wekell, M.M. Detection of sodium channel toxins: Directed cytotoxicity assays of purified ciguatoxins, brevetoxins, saxitoxins, and seafood extracts. J. AOAC Int. 1995, 78, 521–527. [Google Scholar]
- Choi, H.; Engene, N.; Smith, J.E.; Preskitt, L.B.; Gerwick, W.H. Crossbyanols A–D, toxic brominated polyphenyl ethers from the Hawai’ian bloom-forming cyanobacterium Leptolyngbya crossbyana. J. Nat. Prod. 2010, 73, 517–522. [Google Scholar]
- Pereira, A.; Cao, Z.; Murray, T.F.; Gerwick, W.H. Hoiamide A, a sodium channel activator of unusual architecture from a consortium of two papua new Guinea Cyanobacteria. Chem. Biol. 2009, 16, 893–906. [Google Scholar]
- Choi, H.; Pereira, A.R.; Cao, Z.; Shuman, C.F.; Engene, N.; Byrum, T.; Matainaho, T.; Murray, T.F.; Mangoni, A.; Gerwick, W.H. The hoiamides, structurally intriguing neurotoxic lipopeptides from Papua New Guinea marine cyanobacteria. J. Nat. Prod. 2010, 73, 1411–1421. [Google Scholar]
- Malloy, K.L.; Choi, H.; Fiorilla, C.; Valeriote, F.A.; Matainaho, T.; Gerwick, W.H. Hoiamide D, a marine cyanobacteria-derived inhibitor of p53/MDM2 interaction. Bioorg. Med. Chem. Lett. 2012, 22, 683–688. [Google Scholar]
- Wang, L.; Xu, Z.; Ye, T. Total synthesis of hoiamide C. Org. Lett. 2011, 13, 2506–2509. [Google Scholar]
- Philkhana, S.C.; Seetharamsingh, B.; Dangat, Y.B.; Vanka, K.; Reddy, D.S. Synthesis of palmyrolide A and its cis-isomer and mechanistic insight into trans–cis isomerisation of the enamide macrocycle. Chem. Commun. 2013, 49, 3342–3344. [Google Scholar]
- Pereira, A.R.; Cao, Z.; Engene, N.; Soria-Mercado, I.E.; Murray, T.F.; Gerwick, W.H. Palmyrolide A, an unusually stabilized neuroactive macrolide from palmyra atoll cyanobacteria. Org. Lett. 2010, 12, 4490–4493. [Google Scholar]
- Tello-Aburto, R.; Newar, T.D.; Maio, W.A. Evolution of a protecting-group-free total synthesis: Studies en route to the neuroactive marine macrolide (−)-palmyrolide A. J. Org. Chem. 2012, 77, 6271–6289. [Google Scholar]
- Tello-Aburto, R.; Johnson, E.M.; Valdez, C.K.; Maio, W.A. Asymmetric total synthesis and absolute stereochemistry of the neuroactive marine macrolide palmyrolide A. Org Lett. 2012, 14, 2150–2153. [Google Scholar]
- Wadsworth, A.D.; Furkert, D.P.; Sperry, J.; Brimble, M.A. Total synthesis of the initially reported and revised structures of the neuroprotective agent palmyrolide A. Org. Lett. 2012, 14, 5374–5377. [Google Scholar]
- Taniguchi, M.; Nunnery, J.K.; Engene, N.; Esquenazi, E.; Byrum, T.; Dorrestein, P.C.; Gerwick, W.H. Palmyramide A, a cyclic depsipeptide from a palmyra atoll collection of the marine cyanobacterium Lyngbya majuscule. J. Nat. Prod. 2010, 73, 393–398. [Google Scholar]
- Tan, L.T.; Sitachitta, N.; Gerwick, W.H. The guineamides, novel cyclic depsipeptides from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscule. J. Nat. Prod. 2003, 66, 764–771. [Google Scholar]
- Glaser, K.B.; Mayer, A.M.S. A renaissance in marine pharmacology: from preclinical curiosity to clinical reality. Biochem. Pharmacol. 2009, 78, 440–448. [Google Scholar]
- Olivera, B.M.; Cruz, L.J.; de Santos, V.; LeCheminant, G.W.; Griffin, D.; Zeikus, R.; McIntosh, J.M.; Galyean, R.; Varga, J.; Gray, W.R.; et al. Neuronal calcium channel antagonists. Discrimination between calcium channel cubtypes using ω-conotoxin from Conus magus venom. Biochemistry 1987, 26, 2086–2090. [Google Scholar]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar]
- Klotz, U. Ziconotide—a novel neuron-specific calcium channel blocker for the Intrathecal treatment of severe chronic pain—a short review. Int. J. Clin. Pharmacol. Ther. 2006, 44, 478–483. [Google Scholar] [CrossRef]
- Goldberg, Y.P.; MacFarlane, J.; MacDonald, M.L.; Thompson, J.; Dube, M.P.; Mattice, M.; Fraser, R.; Young, C.; Hossain, S.; Pape, T.; et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 2007, 71, 311–319. [Google Scholar] [CrossRef]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar]
- Fertleman, C.R.; Baker, M.D.; Parker, K.A.; Moffatt, S.; Elmslie, F.V.; Abrahamsen, B.; Ostman, J.; Klugbauer, N.; Wood, J.N.; Gardiner, R.M.; et al. SCN9A mutations in paroxysmal extreme pain disorder: Allelic variants underlie distinct channel defects and phenotypes. Neuron 2006, 52, 767–774. [Google Scholar] [CrossRef]
- Han, C.; Rush, A.M.; Dib-Hajj, S.D.; Li, S.; Xu, Z.; Wang, Y.; Tyrrell, L.; Wang, X.; Yang, Y.; Waxman, S.G. Sporadic onset of erythermalgia: A gain-of-function mutation in Nav1.7. Ann. Neurol. 2006, 59, 553–558. [Google Scholar] [CrossRef]
- Ekberg, J.; Jayamanne, A.; Vaughan, C.W.; Aslan, S.; Thomas, L.; Mould, J.; Drinkwater, R.; Baker, M.D.; Abrahamsen, B.; Wood, J.N.; et al. μO-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. Proc. Natl. Acad. Sci. USA 2006, 103, 17030–17035. [Google Scholar]
- Epstein-Barash, H.; Shichor, I.; Kwon, A.H.; Hall, S.; Lawlor, M.W.; Langer, R.; Kohane, D.S. Prolonged duration local anesthesia with minimal toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7125–7130. [Google Scholar]
- Garrido, R.; Laqos, N.; Lattes, K.; Azolas, C.G.; Bocic, G.; Cuneo, A.; Chiong, H.; Jensen, C.; Heríandez, A.; Fernández, C. The Gonyautoxin 2/3 epimers reduces anal tone when injected in the anal sphincter of healthy adults. Biol. Res. 2004, 37, 395–403. [Google Scholar]
- Poh, A.; Tan, K.Y.; Seow-Choen, F. Innovations in chronic anal fissure treatment: A systematic review. World J. Gastrointest. Surg. 2010, 2, 231–241. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, F.; Xu, X.; Li, T.; Liu, Z. Shellfish Toxins Targeting Voltage-Gated Sodium Channels. Mar. Drugs 2013, 11, 4698-4723. https://doi.org/10.3390/md11124698
Zhang F, Xu X, Li T, Liu Z. Shellfish Toxins Targeting Voltage-Gated Sodium Channels. Marine Drugs. 2013; 11(12):4698-4723. https://doi.org/10.3390/md11124698
Chicago/Turabian StyleZhang, Fan, Xunxun Xu, Tingting Li, and Zhonghua Liu. 2013. "Shellfish Toxins Targeting Voltage-Gated Sodium Channels" Marine Drugs 11, no. 12: 4698-4723. https://doi.org/10.3390/md11124698
APA StyleZhang, F., Xu, X., Li, T., & Liu, Z. (2013). Shellfish Toxins Targeting Voltage-Gated Sodium Channels. Marine Drugs, 11(12), 4698-4723. https://doi.org/10.3390/md11124698