Marine-Derived Angiogenesis Inhibitors for Cancer Therapy

Abstract
:1. Introduction
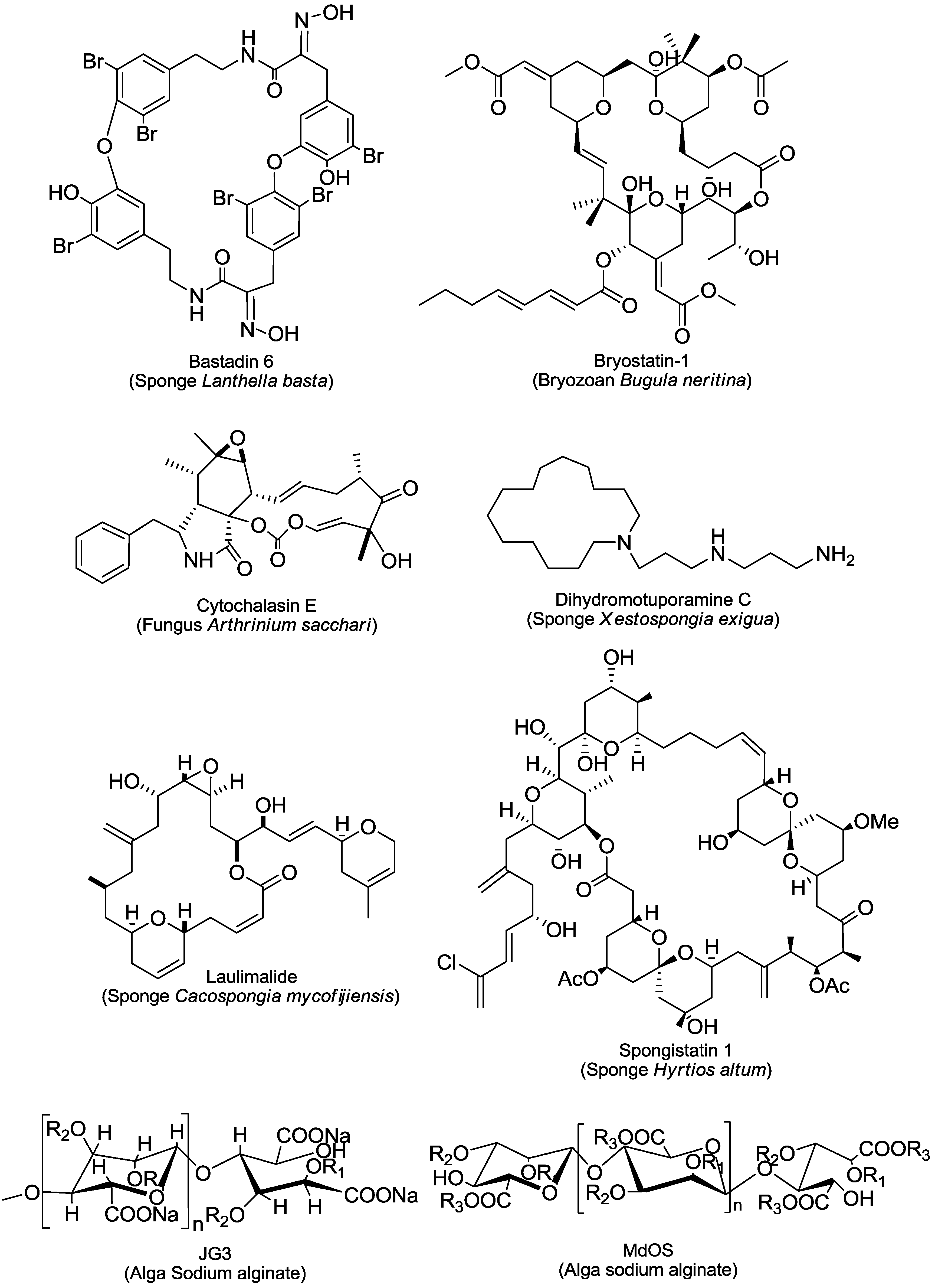
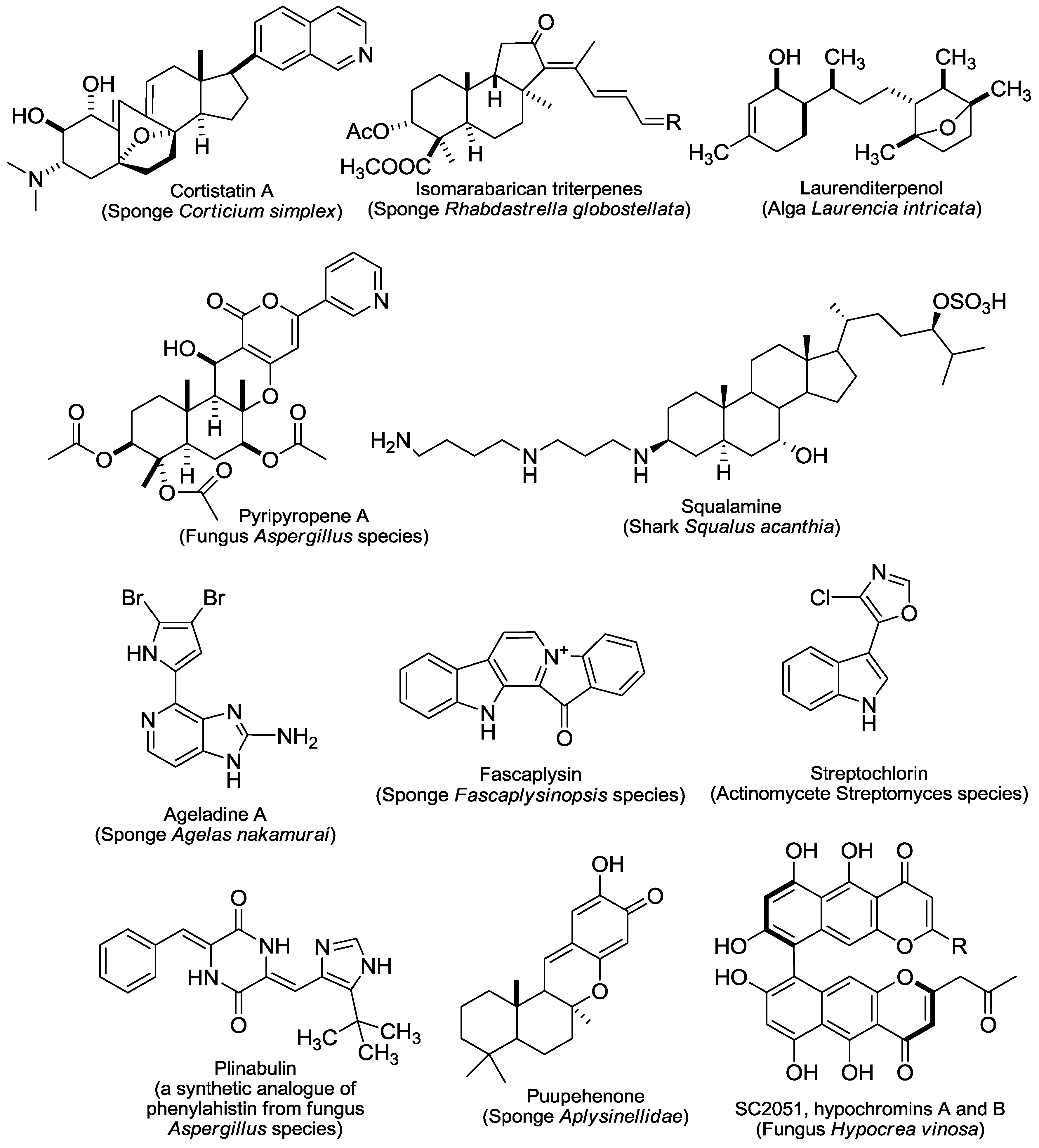
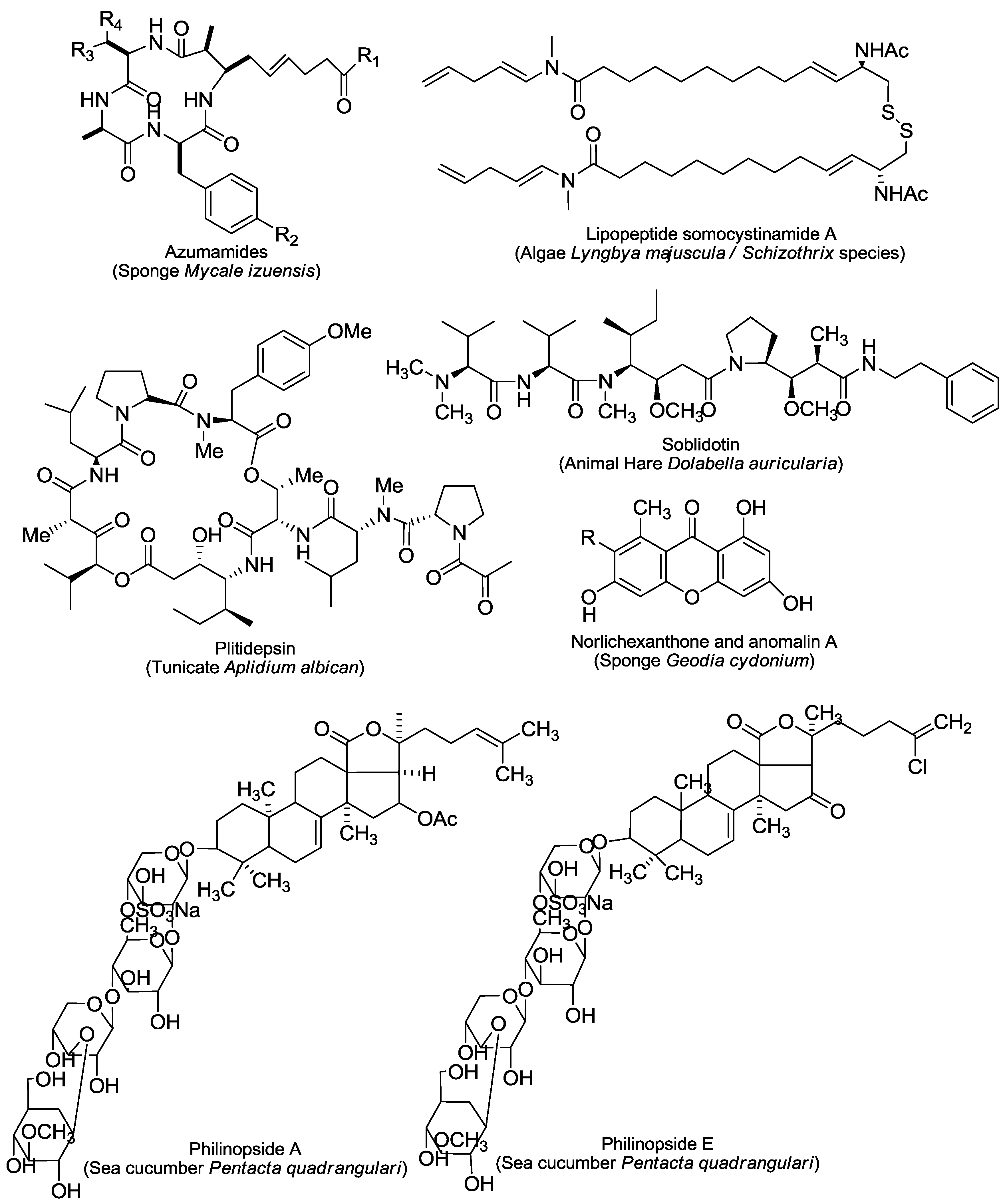
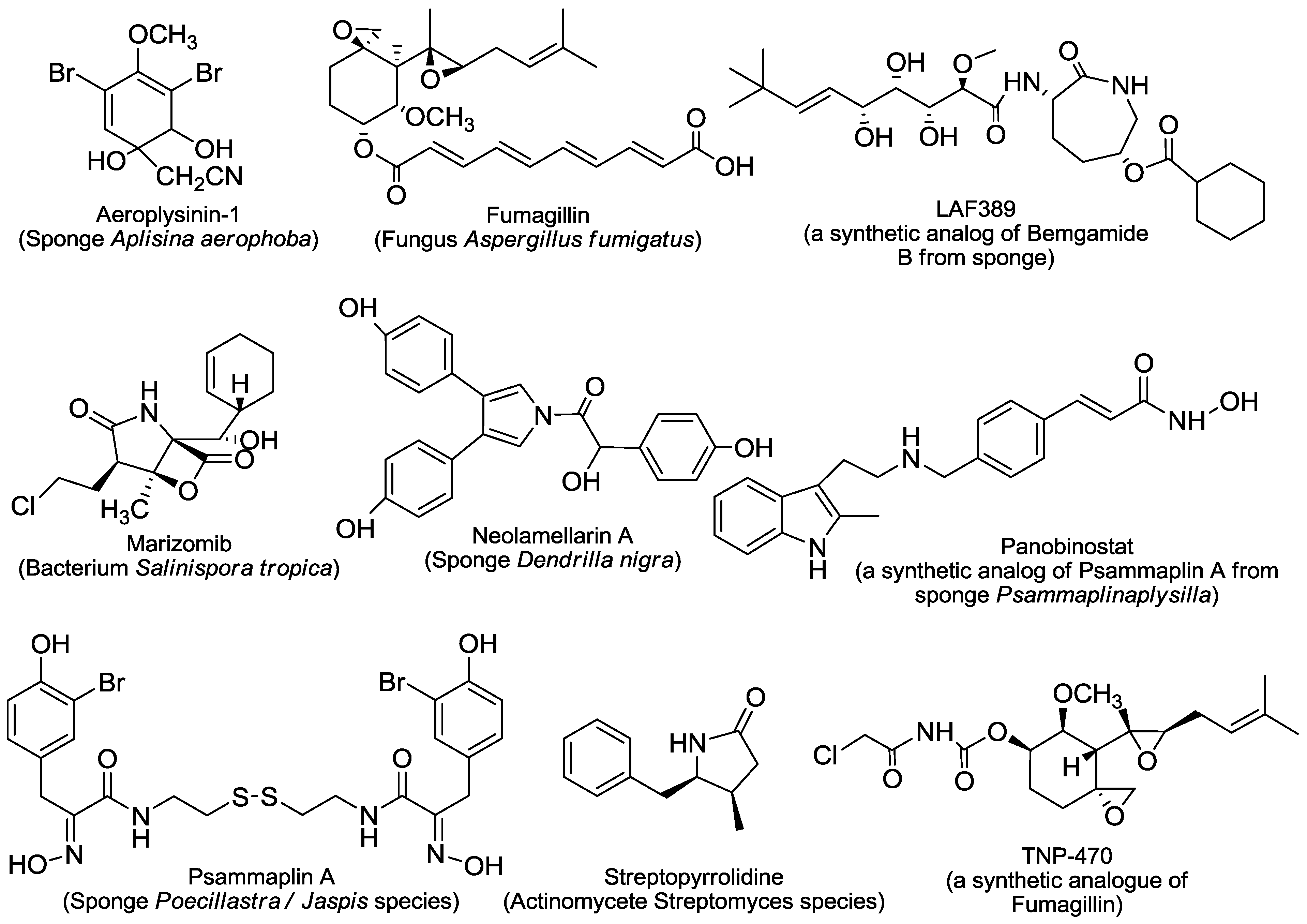
2. An Overview of Marine-Derived Antiangiogenic Agents

 ;hypochromins A, R =
;hypochromins A, R =  ; and hypochromins B, R = CH3.
;hypochromins A, R = ; and hypochromins B, R = CH3.
; and hypochromins B, R = CH3.
;hypochromins A, R = ; and hypochromins B, R = CH3.


2.1. Protein Kinase Modulators
2.2. Cytoskeleton Disturbing Agents

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In vitro assays: |
|
| Ex vivo assays: |
|
| In vivo assays: |
|
2.3. HDAC Inhibitors
2.4. MetAPs Inhibitor
2.5. Others
3. Marine-Derived Protein Kinase Modulators with Antiangiogenic Activity
3.1. S/TK Modulators
3.1.1. The PKC Activator Bryostatin-1
3.1.2. The CDK4 Inhibitor Fascaplysin
3.1.3. Cortistatin A
3.2. TK Inhibitors
3.2.1. Neovastat
3.2.2. Aeroplysinin-1
3.2.3. Philinopside A and Philinopside E
3.2.4. Ageladine A
3.2.5. Saccharides
3.2.6. Anomalin A and Norlichexanthone
3.2.7. SC2051 and hypochromins A and B
4. Clinical Trials of Marine-Derived Angiogenesis Inhibitors for Cancer Therapy
4.1. The PKC Activator Bryostatin-1
4.2. The HDAC Inhibitor Panobinostat
4.3. The Cyclic Depsipeptide Plitidepsin
4.4. The Proteasome Inhibitor Marizomib
4.5. The Tubulin Inhibitor Plinabulin
5. Conclusions
Acknowledgements
Conflict of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Rapisarda, A.; Melillo, G. Role of the VEGF/VEGFR axis in cancer biology and therapy. Adv. Cancer Res. 2012, 114, 237–267. [Google Scholar]
- Siefert, S.A.; Sarkar, R. Matrix metalloproteinases in vascular physiology and disease. Vascular 2012, 20, 210–216. [Google Scholar]
- Yin, S.Q.; Wang, J.J.; Zhang, C.M.; Liu, Z.P. The development of MetAP-2 inhibitors in cancer treatment. Curr. Med. Chem. 2012, 19, 1021–1035. [Google Scholar] [CrossRef]
- Bayless, K.J.; Johnson, G.A. Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J. Vasc. Res. 2011, 48, 369–385. [Google Scholar] [CrossRef]
- Mottet, D.; Castronovo, V. Histone deacetylases: Anti-angiogenic targets in cancer therapy. Curr. Cancer Drug Targets 2010, 10, 898–913. [Google Scholar]
- Miao, Z.H.; Feng, J.M.; Ding, J. Newly discovered angiogenesis inhibitors and their mechanisms of action. Acta Pharmacol. Sin. 2012, 33, 1103–1111. [Google Scholar] [CrossRef]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef]
- Abraham, I.; El Sayed, K.; Chen, Z.S.; Guo, H. Current status on marine products with reversal effect on cancer multidrug resistance. Mar. Drugs 2012, 10, 2312–2321. [Google Scholar] [CrossRef]
- Schwartz, E.L. Antivascular actions of microtubule-binding drugs. Clin. Cancer Res. 2009, 15, 2594–2601. [Google Scholar] [CrossRef]
- Natsume, T.; Watanabe, J.; Tamaoki, S.; Fujio, N.; Miyasaka, K.; Kobayashi, M. Characterization of the interaction of TZT-1027, a potent antitumor agent, with tubulin. Jpn. J. Cancer Res. 2000, 91, 737–747. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Sumikura, M.; Hidaka, K.; Yasui, H.; Kiso, Y.; Yakushiji, F.; Hayashi, Y. Anti-microtubule “plinabulin” chemical probe KPU-244-B3 labeled both α- and β-tubulin. Bioorg. Med. Chem. 2010, 18, 3169–3174. [Google Scholar] [CrossRef]
- Bai, R.; Taylor, G.F.; Cichacz, Z.A.; Herald, C.L.; Kepler, J.A.; Pettit, G.R.; Hamel, E. The spongistatins, potently cytotoxic inhibitors of tubulin polymerization, bind in a distinct region of the vinca domain. Biochemistry 1995, 34, 9714–9721. [Google Scholar]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule stabilizing agents. Cancer Res. 1999, 59, 653–660. [Google Scholar]
- Bennett, M.J.; Barakat, K.; Huzil, J.T.; Tuszynski, J.; Schriemer, D.C. Discovery and characterization of the laulimalide-microtubule binding mode by mass shift perturbation mapping. Chem. Biol. 2010, 17, 725–734. [Google Scholar]
- Watanabe, J.; Natsume, T.; Fujio, N.; Miyasaka, K.; Kobayashi, M. Induction of apoptosis in human cancer cells by TZT-1027, an antimicrotubule agent. Apoptosis 2000, 5, 345–353. [Google Scholar] [CrossRef]
- Honda-Uezono, A.; Kaida, A.; Michi, Y.; Harada, K.; Hayashi, Y.; Hayashi, Y.; Miura, M. Unusual expression of red, fluorescence at M phase induced by anti-microtubule agents in He La cells expressing the fluorescent ubiquitination-based cell cycle indicator (Fucci). Biochem. Biophys. Res. Commun. 2012, 428, 224–229. [Google Scholar]
- Rothmeier, A.S.; Schneiders, U.M.; Wiedmann, R.M.; Ischenko, I.; Bruns, C.J.; Rudy, A.; Zahler, S.; Vollmar, A.M. The marine compound spongistatin 1 targets pancreatic tumor progression and metastasis. Int. J. Cancer 2010, 127, 1096–1105. [Google Scholar] [CrossRef]
- Bennett, M.J.; Chan, G.K.; Rattner, J.B.; Schriemer, D.C. Low-dose laulimalide represents a novel molecular probe for investigating microtubule organization. Cell Cycle 2012, 11, 3045–3054. [Google Scholar] [CrossRef]
- Nicholson, B.; Lloyd, G.K.; Miller, B.R.; Palladino, M.A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S.T.C. NPI-2358 is a tubulin-depolymerizing agent: In vitro evidence for activity as a tumor vascular-disrupting agent. Anticancer Drugs 2006, 17, 25–31. [Google Scholar] [CrossRef]
- Rothmeier, A.S.; Ischenko, I.; Joore, J.; Garczarczyk, D.; Furst, R.; Bruns, C.J.; Vollmar, A.M.; Zahler, S. Investigation of the marine compound spongistatin 1 links the inhibition of PKC α translocation to nonmitotic effects of tubulin antagonism in angiogenesis. FASEB J. 2009, 23, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.Y.; Murtagh, J.; Schwartz, E.L. The microtubule binding drug laulimalide inhibits vascular endothelial growth factor-induced human endothelial cell migration and is synergistic when combined with docetaxel (taxotere). Mol. Pharmacol. 2006, 69, 1207–1215. [Google Scholar]
- Watanabe, J.; Endo, Y.; Shimada, N.; Natsume, T.; Sasaki, T.; Kobayashi, M. Antiangiogenic activity of TZT-1027 (Soblidotin) on chick chorioallantoic membrane and human umbilical vein endothelial cells. In Vivo 2007, 21, 297–304. [Google Scholar]
- Otani, M.; Natsume, T.; Watanabe, J.; Kobayashi, M.; Murakoshi, M.; Mikami, T.; Nakayama, T. TZT-1027, an antimicrotubule agent, attacks tumor vasculature and induces tumor cell death. Jpn. J. Cancer Res. 2000, 91, 837–844. [Google Scholar] [CrossRef]
- Millward, M.; Mainwaring, P.; Mita, A.; Federico, K.; Lloyd, G.K.; Reddinger, N.; Nawrocki, S.; Mita, M.; Spear, M.A. Phase 1 study of the novel vascular disrupting agent plinabulin (NPI-2358) and docetaxel. Invest. New Drugs 2012, 30, 1065–1073. [Google Scholar] [CrossRef]
- Udagawa, T.; Yuan, J.; Panigrahy, D.; Chang, Y.H.; Shah, J.; D’Amato, R.J. Cytochalasin E, an epoxide containing Aspergillus-derived fungal metabolite, inhibits angiogenesis and tumor growth. J. Pharmacol. Exp. Ther. 2000, 294, 421–427. [Google Scholar]
- Duncan, M.D.; Harmon, J.W.; Duncan, K.L.K. Actin disruption inhibits bombesin stimulation of focal adhesion kinase (pp125(FAK)) in prostate carcinoma. J. Surg. Res. 1996, 63, 359–363. [Google Scholar]
- Ikewaki, N.; Yamada, A.; Inoko, H. Depolymerization of actin filament by cytochalasin E induces interleukin-8 production and up-regulates CD54 in the HeLa epithelial cell line. Microbiol. Immunol. 2003, 47, 775–783. [Google Scholar]
- Cox, A.C. Cytochalasin E enhances the protein kinase C-dependent process of secretion. Biochem. Biophys. Res. Commun. 1988, 150, 745–751. [Google Scholar] [CrossRef]
- McHardy, L.M.; Sinotte, R.; Troussard, A.; Sheldon, C.; Church, J.; Williams, D.E.; Andersen, R.J.; Dedhar, S.; Roberge, M.; Roskelley, C.D. The tumor invasion inhibitor dihydromotuporamine C activates RHO, remodels stress fibers and focal adhesions, and stimulates sodium-proton exchange. Cancer Res. 2004, 64, 1468–1474. [Google Scholar] [CrossRef]
- Roskelley, C.D.; Williams, D.E.; McHardy, L.M.; Leong, K.G.; Troussard, A.; Karsan, A.; Andersen, R.J.; Dedhar, S.; Roberge, M. Inhibition of tumor cell invasion and angiogenesis by motuporamines. Cancer Res. 2001, 61, 6788–6794. [Google Scholar]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef]
- Fortunati, N.; Catalano, M.G.; Marano, F.; Mugoni, V.; Pugliese, M.; Bosco, O.; Mainini, F.; Boccuzzi, G. The pan-DAC inhibitor LBH589 is a multi-functional agent in breast cancer cells: Cytotoxic drug and inducer of sodium-iodide symporter (NIS). Breast Cancer Res. Treat. 2010, 124, 667–675. [Google Scholar]
- Qian, D.Z.; Kato, Y.; Shabbeer, S.; Wei, Y.F.; Verheul, H.M.W.; Salumbides, B.; Sanni, T.; Atadja, P.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors: The hydroxamic acid derivative LBH589. Clin. Cancer Res. 2006, 12, 634–642. [Google Scholar] [CrossRef]
- Nakao, Y.; Yoshida, S.; Matsunaga, S.; Shindoh, N.; Terada, Y.; Nagai, K.; Yamashita, J.K.; Ganesan, A.; van Soest, R.W.; Fusetani, N. Azumamides A–E: Histone deacetylase inhibitory cyclic tetrapeptides from the marine sponge Mycale izuensis. Angew. Chem. Int. Ed. Engl. 2006, 45, 7553–7557. [Google Scholar] [CrossRef]
- Nakao, Y.; Narazaki, G.; Hoshino, T.; Maeda, S.; Yoshida, M.; Maejima, H.; Yamashita, J.K. Evaluation of antiangiogenic activity of azumamides by the in vitro vascular organization model using mouse induced pluripotent stem (iPS) cells. Bioorg. Med. Chem. Lett. 2008, 18, 2982–2984. [Google Scholar]
- Pina, I.C.; Gautschi, J.T.; Wang, G.Y.S.; Sanders, M.L.; Schmitz, F.J.; France, D.; Cornell-Kennon, S.; Sambucetti, L.C.; Remiszewski, S.W.; Perez, L.B.; et al. Psammaplins from the sponge Pseudoceratina purpurea: Inhibition of both histone deacetylase and DNA methyltransferase. J. Org. Chem. 2003, 68, 3866–3873. [Google Scholar] [CrossRef]
- Shim, J.S.; Lee, H.S.; Shin, J.; Kwon, H.J. Psammaplin A, a marine natural product, inhibits aminopeptidase N and suppresses angiogenesis in vitro. Cancer Lett. 2004, 203, 163–169. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Jung, J.H.; Na, Y.J.; Kim, H.S. A natural histone deacetylase inhibitor, Psammaplin A, induces cell cycle arrest and apoptosis in human endometrial cancer cells. Gynecol. Oncol. 2008, 108, 27–33. [Google Scholar] [CrossRef]
- Kim, D.H.; Shin, J.; Kwon, H.J. Psammaplin A is a natural prodrug that inhibits class I histone deacetylase. Exp. Mol. Med. 2007, 39, 47–55. [Google Scholar] [CrossRef]
- Mauriz, J.L.; Martin-Renedo, J.; Garcia-Palomo, A.; Tunon, M.J.; Gonzalez-Gallego, J. Methionine aminopeptidases as potential targets for treatment of gastrointestinal cancers and other tumors. Curr. Drug Targets 2010, 11, 1430–1448. [Google Scholar] [CrossRef]
- Datta, B. Roles of P67/MetAP2 as a tumor suppressor. Biochim. Biophys. Acta 2009, 1796, 281–292. [Google Scholar]
- Sato, Y. Role of aminopeptidase in angiogenesis. Biol. Pharm. Bull. 2004, 27, 772–776. [Google Scholar] [CrossRef]
- Hines, J.; Ju, R.; Dutschman, G.E.; Cheng, Y.C.; Crews, C.M. Reversal of TNP-470-induced endothelial cell growth arrest by guanine and guanine nucleosides. J. Pharmacol. Exp. Ther. 2010, 334, 729–738. [Google Scholar] [CrossRef]
- Chen, G.J.; Weylie, B.; Hu, C.; Zhu, J.; Forough, R. FGFR1/PI3K/AKT signaling pathway is a novel target for antiangiogenic effects of the cancer drug fumagillin (TNP-470). J. Cell. Biochem. 2007, 101, 1492–1504. [Google Scholar]
- Xu, W.; Lu, J.P.; Ye, Q.Z. Structural analysis of bengamide derivatives as inhibitors of methionine aminopeptidases. J. Med. Chem. 2012, 55, 8021–8027. [Google Scholar] [CrossRef]
- Kinder, F.R.; Versace, R.W.; Bair, K.W.; Bontempo, J.M.; Cesarz, D.; Chen, S.; Crews, P.; Czuchta, A.M.; Jagoe, C.T.; Mou, Y.; et al. Synthesis and antitumor activity of ester-modified analogues of bengamide B. J. Med. Chem. 2001, 44, 3692–3699. [Google Scholar] [CrossRef]
- Towbin, H.; Bair, K.W.; DeCaprio, J.A.; Eck, M.J.; Kim, S.; Kinder, F.R.; Morollo, A.; Mueller, D.R.; Schindler, P.; Song, H.K.; et al. Proteomics-based target identification: Bengamides as a new class of methionine aminopeptidase inhibitors. J. Biol. Chem. 2003, 278, 52964–52971. [Google Scholar] [CrossRef]
- Dumez, H.; Gall, H.; Capdeville, R.; Dutreix, C.; van Oosterom, A.T.; Giaccone, G. A phase I and pharmacokinetic study of LAF389 administered to patients with advanced cancer. Anticancer Drugs 2007, 18, 219–225. [Google Scholar] [CrossRef]
- Castro, M.E.; Gonzalez-Iriarte, M.; Barrero, A.F.; Salvador-Tormo, N.; Munoz-Chapuli, R.; Medina, M.A.; Quesada, A.R. Study of puupehenone and related compounds as inhibitors of angiogenesis. Int. J. Cancer 2004, 110, 31–38. [Google Scholar] [CrossRef]
- Choi, I.K.; Shin, H.J.; Lee, H.S.; Kwon, H.J. Streptochlorin, a marine natural product, inhibits NF-κB activation and suppresses angiogenesis in vitro. J. Microbiol. Biotechnol. 2007, 17, 1338–1343. [Google Scholar]
- Wrasidlo, W.; Mielgo, A.; Torres, V.A.; Barbero, S.; Stoletov, K.; Suyama, T.L.; Klemke, R.L.; Gerwick, W.H.; Carson, D.A.; Stupack, D.G. The marine lipopeptide somocystinamide A triggers apoptosis via caspase 8. Proc. Natl. Acad. Sci. USA 2008, 105, 2313–2318. [Google Scholar] [CrossRef]
- Aoki, S.; Cho, S.; Ono, M.; Kuwano, T.; Nakao, S.; Kuwano, M.; Nakagawa, S.; Gao, J.Q.; Mayumi, T.; Shibuya, M.; et al. Bastadin 6, a spongean brominated tyrosine derivative, inhibits tumor angiogenesis by inducing selective apoptosis to endothelial cells. Anticancer Drugs 2006, 17, 269–278. [Google Scholar] [CrossRef]
- Hayashi, A.; Arai, M.; Fujita, M.; Kobayashi, M. Pyripyropenes, fungal sesquiterpenes conjugated with α-pyrone and pyridine moieties, exhibits anti-angiogenic activity against human umbilical vein endothelial cells. Biol. Pharm. Bull. 2009, 32, 1261–1265. [Google Scholar] [CrossRef]
- Aoki, S.; Sanagawa, M.; Watanabe, Y.; Setiawan, A.; Arai, M.; Kobayashi, M. Novel isomarabarican triterpenes, exhibiting selective anti-proliferative activity against vascular endothelial cells, from marine sponge Rhabdastrella globostellata. Bioorg. Med. Chem. 2007, 15, 4818–4828. [Google Scholar] [CrossRef]
- Shin, H.J.; Kim, T.S.; Lee, H.S.; Park, J.Y.; Choi, I.K.; Kwon, H.J. Streptopyrrolidine, an angiogenesis inhibitor from a marine-derived Streptomyces sp. KORDI-3973. Phytochemistry 2008, 69, 2363–2366. [Google Scholar]
- Dias, P.F.; Siqueira, J.M., Jr.; Maraschin, M.; Ferreira, A.G.; Gagliardi, A.R.; Ribeiro-do-Valle, R.M. A polysaccharide isolated from the brown seaweed S argassum stenophyllum exerts antivasculogenic effects evidenced by modified morphogenesis. Microvasc. Res. 2008, 75, 34–44. [Google Scholar] [CrossRef]
- Matsubara, K.; Mori, M.; Matsumoto, H.; Hori, K.; Miyazawa, K. Antiangiogenic properties of a sulfated galactan isolated from a marine green alga, Codium cylindricum. J. Appl. Phycol. 2003, 15, 87–90. [Google Scholar] [CrossRef]
- Liu, F.; Wang, J.; Chang, A.K.; Liu, B.; Yang, L.L.; Li, Q.M.; Wang, P.S.; Zou, X.Y. Fucoidan extract derived from Undaria pinnatifida inhibits angiogenesis by human umbilical vein endothelial cells. Phytomedicine 2012, 19, 797–803. [Google Scholar] [CrossRef]
- Xue, M.; Ge, Y.; Zhang, J.; Wang, Q.; Hou, L.; Liu, Y.; Sun, L.; Li, Q. Anticancer properties and mechanisms of fucoidan on mouse breast cancer in vitro and in vivo. PLoS One 2012, 7, e43483. [Google Scholar]
- Chittiboyina, A.G.; Kumar, G.M.; Carvalho, P.B.; Liu, Y.; Zhou, Y.D.; Nagle, D.G.; Avery, M.A. Total synthesis and absolute configuration of laurenditerpenol: A hypoxia inducible factor-1 activation inhibitor. J. Med. Chem. 2007, 50, 6299–6302. [Google Scholar] [CrossRef]
- Arafeh, K.M.; Ullah, N. Synthesis of neolamellarin A, an inhibitor of hypoxia-inducible factor-1. Nat. Prod. Commun. 2009, 4, 925–926. [Google Scholar]
- Taraboletti, G.; Poli, M.; Dossi, R.; Manenti, L.; Borsotti, P.; Faircloth, G.T.; Broggini, M.; D’Incalci, M.; Ribatti, D.; Giavazzi, R. Antiangiogenic activity of aplidine, a new agent of marine origin. Br. J. Cancer 2004, 90, 2418–2424. [Google Scholar]
- Straight, A.M.; Oakley, K.; Moores, R.; Bauer, A.J.; Patel, A.; Tuttle, R.M.; Jimeno, J.; Francis, G.L. Aplidin reduces growth of anaplastic thyroid cancer xenografts and the expression of several angiogenic genes. Cancer Chemother. Pharmacol. 2006, 57, 7–14. [Google Scholar] [CrossRef]
- Caers, J.; Menu, E.; de Raeve, H.; Lepage, D.; van Valckenborgh, E.; van Camp, B.; Alvarez, E.; Vanderkerken, K. Antitumour and antiangiogenic effects of Aplidin in the 5TMM syngeneic models of multiple myeloma. Br. J. Cancer 2008, 98, 1966–1974. [Google Scholar]
- Sills, A.K.; Williams, J.I.; Tyler, B.M.; Epstein, D.S.; Sipos, E.P.; Davis, J.D.; McLane, M.P.; Pitchford, S.; Cheshire, K.; Cannon, F.H.; et al. Squalamine inhibits angiogenesis and solid tumor growth in vivo and perturbs embryonic vasculature. Cancer Res. 1998, 58, 2784–2792. [Google Scholar]
- Ciulla, T.A.; Criswell, M.H.; Danis, R.P.; Williams, J.I.; McLane, M.P.; Holroyd, K.J. Squalamine lactate reduces choroidal neovascularization in a laser-injury model in the rat. Retina 2003, 23, 808–814. [Google Scholar] [CrossRef]
- Hao, D.; Hammond, L.A.; Eckhardt, S.G.; Patnaik, A.; Takimoto, C.H.; Schwartz, G.H.; Goetz, A.D.; Tolcher, A.W.; McCreery, H.A.; Mamun, K.; et al. A phase I and pharmacokinetic study of squalamine, an aminosterol angiogenesis inhibitor. Clin. Cancer Res. 2003, 9, 2465–2471. [Google Scholar]
- Walker, B.T.; Houston, T.A. Squalamine and its derivatives as potential antitubercular compounds. Tuberculosis 2012, 93, 102–103. [Google Scholar] [CrossRef]
- Hraiech, S.; Bregeon, F.; Brunel, J.M.; Rolain, J.M.; Lepidi, H.; Andrieu, V.; Raoult, D.; Papazian, L.; Roch, A. Antibacterial efficacy of inhaled squalamine in a rat model of chronic Pseudomonas aeruginosa pneumonia. J. Antimicrob. Chemother. 2012, 67, 2452–2458. [Google Scholar] [CrossRef]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew. Chem. Int. Ed. Engl. 2003, 42, 355–357. [Google Scholar]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar]
- Ahn, K.S.; Sethi, G.; Chao, T.H.; Neuteboom, S.T.C.; Chaturvedi, M.M.; Palladino, M.A.; Younes, A.; Aggarwal, B.B. Salinosporamide A (NPI-0052) potentiates apoptosis, suppresses osteoclastogenesis, and inhibits invasion through down-modulation of NF-κB-regulated gene products. Blood 2007, 110, 2286–2295. [Google Scholar] [CrossRef]
- Zhu, K.Y.; Chan, W.; Heymach, J.; Wilkinson, M.; McConkey, D.J. Control of HIF-1α expression by eIF2α phosphorylation-mediated translational repression. Cancer Res. 2009, 69, 1836–1843. [Google Scholar] [CrossRef]
- Irie, K.; Yanagita, R.C.; Nakagawa, Y. Challenges to the development of bryostatin-type anticancer drugs based on the activation mechanism of protein kinase Cδ. Med. Res. Rev. 2012, 32, 518–535. [Google Scholar] [CrossRef]
- Kazanietz, M.G.; Lewin, N.E.; Gao, F.; Pettit, G.R.; Blumberg, P.M. Binding of [26-3H]bryostatin 1 and analogs to calcium-dependent and calcium-independent protein kinase C isozymes. Mol. Pharmacol. 1994, 46, 374–379. [Google Scholar]
- Szallasi, Z.; Denning, M.F.; Smith, C.B.; Dlugosz, A.A.; Yuspa, S.H.; Pettit, G.R.; Blumberg, P.M. Bryostatin 1 protects protein kinase C-δ from down-regulation in mouse keratinocytes in parallel with its inhibition of phorbol ester-induced differentiation. Mol. Pharmacol. 1994, 46, 840–850. [Google Scholar]
- Szallasi, Z.; Smith, C.B.; Pettit, G.R.; Blumberg, P.M. Differential regulation of protein kinase C isozymes by bryostatin 1 and phorbol 12-myristate 13-acetate in NIH 3T3 fibroblasts. J. Biol. Chem. 1994, 269, 2118–2124. [Google Scholar]
- Nezhat, F.; Wadler, S.; Muggia, F.; Mandeli, J.; Goldberg, G.; Rahaman, J.; Runowicz, C.; Murgo, A.J.; Gardner, G.J. Phase II trial of the combination of bryostatin-1 and cisplatin in advanced or recurrent carcinoma of the cervix: A New York Gynecologic Oncology Group study. Gynecol. Oncol. 2004, 93, 144–148. [Google Scholar] [CrossRef]
- Mackay, H.J.; Twelves, C.J. Targeting the protein kinase C family: Are we there yet? Nat. Rev. Cancer 2007, 7, 554–562. [Google Scholar] [CrossRef]
- Shafiq, M.I.; Steinbrecher, T.; Schmid, R. FASCAPLYSIN as a specific inhibitor for CDK4: Insights from molecular modelling. PLoS One 2012, 7, e42612. [Google Scholar]
- Zheng, Y.L.; Lu, X.L.; Lin, J.; Chen, H.M.; Yan, X.J.; Wang, F.; Xu, W.F. Direct effects of fascaplysin on human umbilical vein endothelial cells attributing the anti-angiogenesis activity. Biomed. Pharmacother. 2010, 64, 527–533. [Google Scholar] [CrossRef]
- Yan, X.J.; Chen, H.M.; Lu, X.L.; Wang, F.; Xu, W.F.; Jin, H.X.; Zhu, P. Fascaplysin exert anti-tumor effects through apoptotic and anti-angiogenesis pathways in sarcoma mice model. Eur. J. Pharm. Sci. 2011, 43, 251–259. [Google Scholar]
- Lin, J.; Yan, X.J.; Chen, H.M. Fascaplysin, a selective CDK4 inhibitor, exhibit anti-angiogenic activity in vitro and in vivo. Cancer Chemother. Pharmacol. 2007, 59, 439–445. [Google Scholar] [CrossRef]
- Aoki, S.; Watanabe, Y.; Sanagawa, M.; Setiawan, A.; Kotoku, N.; Kobayashi, M. Cortistatins A, B, C, and D, anti-angiogenic steroidal alkaloids, from the marine sponge Corticium simplex. J. Am. Chem. Soc. 2006, 128, 3148–3149. [Google Scholar] [CrossRef]
- Aoki, S.; Watanabe, Y.; Tanabe, D.; Arai, M.; Suna, H.; Miyamoto, K.; Tsujibo, H.; Tsujikawa, K.; Yamamoto, H.; Kobayashi, M. Structure-activity relationship and biological property of cortistatins, anti-angiogenic spongean steroidal alkaloids. Bioorg. Med. Chem. 2007, 15, 6758–6762. [Google Scholar] [CrossRef]
- Cee, V.J.; Chen, D.Y.K.; Lee, M.R.; Nicolaou, K.C. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew. Chem. Int. Ed. Engl. 2009, 48, 8952–8957. [Google Scholar] [CrossRef]
- Dupont, E.; Falardeau, P.; Mousa, S.A.; Dimitriadou, V.; Pepin, M.C.; Wang, T.Q.; Alaoui-Jamali, M.A. Antiangiogenic and antimetastatic properties of Neovastat (AE-941), an orally active extract derived from cartilage tissue. Clin. Exp. Metastas 2002, 19, 145–153. [Google Scholar] [CrossRef]
- Beliveau, R.; Gingras, D.; Kruger, E.A.; Lamy, S.; Sirois, P.; Tranqui, L.; Baffert, F.; Beaulieu, E.; Dimitriadou, V.; Pepin, M.C.; et al. The antiangiogenic agent Neovastat (AE-941) inhibits vascular endothelial growth factor-mediated biological effects. Clin. Cancer Res. 2002, 8, 1242–1250. [Google Scholar]
- Gingras, D.; Renaud, A.; Mousseau, N.; Beaulieu, E.; Kachra, Z.; Beliveau, R. Matrix proteinase inhibition by AE-941, a multifunctional antiangiogenic compound. Anticancer Res. 2001, 21, 145–155. [Google Scholar]
- Gingras, D.; Labelle, D.; Nyalendo, C.; Boivin, D.; Demeule, M.; Barthomeuf, C.; Beliveau, R. The antiangiogenic agent Neovastat (AE-941) stimulates tissue plasminogen activator activity. Invest. New Drugs 2004, 22, 17–26. [Google Scholar] [CrossRef]
- Boivin, D.; Gendron, S.; Beaulieu, E.; Gingras, D.; Beliveau, R. The antiangiogenic agent Neovastat (AE-941) induces endothelial cell apoptosis. Mol. Cancer Ther. 2002, 1, 795–802. [Google Scholar]
- Falardeau, P.; Champagne, P.; Poyet, P.; Hariton, C.; Dupont, E. Neovastat, a naturally occurring multifunctional antiangiogenic drug, in phase III clinical trials. Semin. Oncol. 2001, 28, 620–625. [Google Scholar] [CrossRef]
- Lu, C.; Lee, J.J.; Komaki, R.; Herbst, R.S.; Feng, L.; Evans, W.K.; Choy, H.; Desjardins, P.; Esparaz, B.T.; Truong, M.T.; et al. Chemoradiotherapy with or without AE-941 in stage III non-small cell lung cancer: A randomized phase III trial. J. Natl. Cancer Inst. 2010, 102, 859–865. [Google Scholar] [CrossRef]
- Escudier, B.; Choueiri, T.K.; Oudard, S.; Szczylik, C.; Negrier, S.; Ravaud, A.; Chevreau, C.; Venner, P.; Champagne, P.; Croteau, D.; et al. Prognostic factors of metastatic renal cell carcinoma after failure of immunotherapy: New paradigm from a large phase III trial with shark cartilage extract AE 941. J. Urol. 2007, 178, 1901–1905. [Google Scholar] [CrossRef]
- Sharma, M.R.; Karrison, T.G.; Jin, Y.; Bies, R.R.; Maitland, M.L.; Stadler, W.M.; Ratain, M.J. Resampling phase III data to assess phase II trial designs and endpoints. Clin. Cancer Res. 2012, 18, 2309–2315. [Google Scholar] [CrossRef]
- Alifrangis, C.; Stebbing, J. Shark cartilage: Has the popularisation of science failed? Lancet Oncol. 2012, 13, 22–22. [Google Scholar] [CrossRef]
- Martinez-Poveda, B.; Rodriguez-Nieto, S.; Garcia-Caballero, M.; Medina, M.A.; Quesada, A.R. The antiangiogenic compound aeroplysinin-1 induces apoptosis in endothelial cells by activating the mitochondrial pathway. Mar. Drugs 2012, 10, 2033–2046. [Google Scholar] [CrossRef]
- Hinterding, K.; Knebel, A.; Herrlich, P.; Waldmann, H. Synthesis and biological evaluation of aeroplysinin analogues: A new class of receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. 1998, 6, 1153–1162. [Google Scholar] [CrossRef]
- Sallam, A.A.; Ramasahayam, S.; Meyer, S.A.; Sayed, K.A. Design, synthesis, and biological evaluation of dibromotyrosine analogues inspired by marine natural products as inhibitors of human prostate cancer proliferation, invasion, and migration. Bioorg. Med. Chem. 2010, 18, 7446–7457. [Google Scholar] [CrossRef]
- Tong, Y.G.; Zhang, X.W.; Tian, F.; Yi, Y.H.; Xu, Q.Z.; Li, L.; Tong, L.J.; Lin, L.P.; Ding, J. Philinopside A, a novel marine-derived compound possessing dual anti-angiogenic and anti-tumor effects. Int. J. Cancer 2005, 114, 843–853. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, X.W.; Tong, Y.G.; Yi, Y.; Zhang, S.L.; Li, L.; Sun, P.; Lin, L.P.; Ding, J. PE, a new sulfated saponin from sea cucumber, exhibits anti-angiogenic and anti-tumor activities in vitro and in vivo. Cancer Biol. Ther. 2005, 4, 874–882. [Google Scholar] [CrossRef]
- Shengule, S.R.; Loa-Kum-Cheung, W.L.; Parish, C.R.; Blairvacq, M.; Meijer, L.; Nakao, Y.; Karuso, P. A one-pot synthesis and biological activity of ageladine a and analogues. J. Med. Chem. 2011, 54, 2492–2503. [Google Scholar]
- Ando, N.; Terashima, S. Synthesis and matrix metalloproteinase (MMP)-12 inhibitory activity of ageladine A and its analogs. Bioorg. Med. Chem. Lett. 2007, 17, 4495–4499. [Google Scholar] [CrossRef]
- Meketa, M.L.; Weinreb, S.M.; Nakao, Y.; Fusetani, N. Application of a 6pi-1-azatriene electrocyclization strategy to the total synthesis of the marine sponge metabolite ageladine A and biological evaluation of synthetic analogues. J. Org. Chem. 2007, 72, 4892–4899. [Google Scholar]
- Shengule, S.R.; Karuso, P. Concise total synthesis of the marine natural product ageladine A. Org. Lett. 2006, 8, 4083–4084. [Google Scholar] [CrossRef]
- Meketa, M.L.; Weinreb, S.M. Total synthesis of ageladine A, an angiogenesis inhibitor from the marine sponge Agelas nakamurai. Org. Lett. 2006, 8, 1443–1446. [Google Scholar] [CrossRef]
- Fujita, M.; Nakao, Y.; Matsunaga, S.; Seiki, M.; Itoh, Y.; Yamashita, J.; van Soest, R.W.M.; Fusetani, N. Bioactive marine metabolites, Part 124. Ageladine A: An antiangiogenic matrixmetalloproteinase inhibitor from the marine sponge Agelas nakamurai. J. Am. Chem. Soc. 2003, 125, 15700–15701. [Google Scholar] [CrossRef]
- Obermann, D.; Bickmeyer, U.; Wagele, H. Incorporated nematocysts in Aeolidiella stephanieae (Gastropoda, Opisthobranchia, Aeolidoidea) mature by acidification shown by the pH sensitive fluorescing alkaloid Ageladine A. Toxicon 2012, 60, 1108–1116. [Google Scholar] [CrossRef]
- Bickmeyer, U. The alkaloid Ageladine A, originally isolated from marine sponges, used for pH-sensitive imaging of transparent marine animals. Mar. Drugs 2012, 10, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.J.; Liu, H.Y.; Chen, Y.; Xin, X.L.; Li, J.; Hou, Y.T.; Zhang, Z.H.; Zhang, X.W.; Me, C.Y.; Geng, M.Y.; et al. Oligomannurarate sulfate, a novel heparanase inhibitor simultaneously targeting basic fibroblast growth factor, combats tumor angiogenesis and metastasis. Cancer Res. 2006, 66, 8779–8787. [Google Scholar] [CrossRef]
- Ma, J.G.; Xin, X.L.; Meng, L.H.; Tong, L.J.; Lin, L.P.; Geng, M.Y.; Ding, J. The marine-derived oligosaccharide sulfate (MdOS), a novel multiple tyrosine kinase inhibitor, combats tumor angiogenesis both in vitro and in vivo. PLoS One 2008, 3, e3774. [Google Scholar]
- Chabut, D.; Fischer, A.M.; Colliec-Jouault, S.; Laurendeau, I.; Matou, S.; Le Bonniec, B.; Helley, D. Low molecular weight fucoidan and heparin enhance the basic fibroblast growth factor-induced tube formation of endothelial cells through heparan sulfate-dependent α6 overexpression. Mol. Pharmacol. 2003, 64, 696–702. [Google Scholar]
- Lake, A.C.; Vassy, R.; Di Benedetto, M.; Lavigne, D.; Le Visage, C.; Perret, G.Y.; Letourneur, D. Low molecular weight fucoidan increases VEGF(165)-induced endothelial cell migration by enhancing VEGF(165) binding to VEGFR-2 and NRP1. J. Biol. Chem. 2006, 281, 37844–37852. [Google Scholar]
- Luyt, C.E.; Meddahi-Pelle, A.; Ho-Tin-Noe, B.; Colliec-Jouault, S.; Guezennec, J.; Louedec, L.; Prats, H.E.; Jacob, M.P.; Osborne-Pellegrin, M.; Letourneur, D.; et al. Low-molecular-weight fucoidan promotes therapeutic revascularization in a rat model of critical hindlimb ischemia. J. Pharmacol. Exp. Ther. 2003, 305, 24–30. [Google Scholar] [CrossRef]
- Matsubara, K.; Xue, C.; Zhao, X.; Mori, M.; Sugawara, T.; Hirata, T. Effects of middle molecular weight fucoidans on in vitro and ex vivo angiogenesis of endothelial cells. Int. J. Mol. Med. 2005, 15, 695–699. [Google Scholar]
- Cumashi, A.; Ushakova, N.A.; Preobrazhenskaya, M.E.; D’Incecco, A.; Piccoli, A.; Totani, L.; Tinari, N.; Morozevich, G.E.; Berman, A.E.; Bilan, M.I.; et al. A comparative study of the anti-inflammatory, anticoagulant, antiangiogenic, and antiadhesive activities of nine different fucoidans from brown seaweeds. Glycobiology 2007, 17, 541–552. [Google Scholar]
- Soeda, S.; Kozako, T.; Iwata, K.; Shimeno, H. Oversulfated fucoidan inhibits the basic fibroblast growth factor-induced tube formation by human umbilical vein endothelial cells: Its possible mechanism of action. Biochim. Biophys. Acta 2000, 1497, 127–134. [Google Scholar] [CrossRef]
- Koyanagi, S.; Tanigawa, N.; Nakagawa, H.; Soeda, S.; Shimeno, H. Oversulfation of fucoidan enhances its anti-angiogenic and antitumor activities. Biochem. Pharmacol. 2003, 65, 173–179. [Google Scholar]
- Dias, P.F.; Siqueira, J.M.; Vendruscolo, L.F.; Neiva, T.D.; Gagliardi, A.R.; Maraschin, M.; Ribeiro-do-Valle, R.M. Antiangiogenic and antitumoral properties of a polysaccharide isolated from the seaweed Sargassum stenophyllum. Cancer Chemother. Pharmacol. 2005, 56, 436–446. [Google Scholar] [CrossRef]
- Tang, X.L.; Li, J.; Xin, X.L.; Geng, M.Y. A new marine-derived sulfated polysaccharide from brown alga suppresses tumor metastasis both in vitro and in vivo. Cancer Biol. Ther. 2006, 5, 1474–1480. [Google Scholar]
- Abdel-Lateff, A.; Klemke, C.; Konig, G.M.; Wright, A.D. Two new xanthone derivatives from the algicolous marine fungus Wardomyces anomalus. J. Nat. Prod. 2003, 66, 706–708. [Google Scholar] [CrossRef]
- Ebada, S.S.; Schulz, B.; Wray, V.; Totzke, F.; Kubbutat, M.H.G.; Muller, W.E.G.; Hamacher, A.; Kassack, M.U.; Lin, W.H.; Proksch, P. Arthrinins A–D: Novel diterpenoids and further constituents from the sponge derived fungus Arthrinium sp. Bioorg. Med. Chem. 2011, 19, 4644–4651. [Google Scholar] [CrossRef]
- Ohkawa, Y.; Miki, K.; Suzuki, T.; Nishio, K.; Sugita, T.; Kinoshita, K.; Takahashi, K.; Koyama, K. Antiangiogenic metabolites from a marine-derived fungus, Hypocrea vinosa. J. Nat. Prod. 2010, 73, 579–582. [Google Scholar] [CrossRef]
- Patel, S.; Keohan, M.L.; Saif, M.W.; Rushing, D.; Baez, L.; Feit, K.; DeJager, R.; Anderson, S. Phase II study of intravenous TZT-1027 in patients with advanced or metastatic soft-tissue sarcomas with prior exposure to anthracycline-based chemotherapy. Cancer 2006, 107, 2881–2887. [Google Scholar] [CrossRef]
- Riely, G.J.; Gadgeel, S.; Rothman, I.; Saidman, B.; Sabbath, K.; Feit, K.; Kris, M.G.; Rizvi, N.A. A phase 2 study of TZT-1027, administered weekly to patients with advanced non-small cell lung cancer following treatment with platinum-based chemotherapy. Lung Cancer 2007, 55, 181–185. [Google Scholar] [CrossRef]
- Logothetis, C.J.; Wu, K.K.; Finn, L.D.; Daliani, D.; Figg, W.; Ghaddar, H.; Gutterman, J.U. Phase I trial of the angiogenesis inhibitor TNP-470 for progressive androgen-independent prostate cancer. Clin. Cancer Res. 2001, 7, 1198–1203. [Google Scholar]
- Folkman, J. Angiogenesis. Annu. Rev. Med. 2006, 57, 1–18. [Google Scholar] [CrossRef]
- Herbst, R.S.; Hammond, L.A.; Carbone, D.P.; Tran, H.T.; Holroyd, K.J.; Desai, A.; Williams, J.I.; Bekele, B.N.; Hait, H.; Allgood, V.; et al. A phase I/IIA trial of continuous five-day infusion of squalamine lactate (MSI-1256F) plus carboplatin and paclitaxel in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2003, 9, 4108–4115. [Google Scholar]
- Varterasian, M.L.; Mohammad, R.M.; Shurafa, M.S.; Hulburd, K.; Pemberton, P.A.; Rodriguez, D.H.; Spadoni, V.; Eilender, D.S.; Murgo, A.; Wall, N.; et al. Phase II trial of bryostatin 1 in patients with relapsed low-grade non-Hodgkin’s lymphoma and chronic lymphocytic leukemia. Clin. Cancer Res. 2000, 6, 825–828. [Google Scholar]
- Pagliaro, L.; Daliani, D.; Amato, R.; Tu, S.M.; Jones, D.; Smith, T.; Logothetis, C.; Millikan, R. A phase II trial of bryostatin-1 for patients with metastatic renal cell carcinoma. Cancer 2000, 89, 615–618. [Google Scholar] [CrossRef]
- Zonder, J.A.; Shields, A.F.; Zalupski, M.; Chaplen, R.; Heilbrun, L.K.; Arlauskas, P.; Philip, P.A. A phase II trial of bryostatin 1 in the treatment of metastatic colorectal cancer. Clin. Cancer Res. 2001, 7, 38–42. [Google Scholar]
- Blackhall, F.H.; Ranson, M.; Radford, J.A.; Hancock, B.W.; Soukop, M.; McGown, A.T.; Robbins, A.; Halbert, G.; Jayson, G.C.; Comm, C.R.; et al. A phase II trial of bryostatin 1 in patients with non-Hodgkin’s lymphoma. Br. J. Cancer 2001, 84, 465–469. [Google Scholar] [CrossRef]
- Bedikian, A.Y.; Plager, C.; Stewart, J.R.; O’Brian, C.A.; Herdman, S.K.; Ross, M.; Papadopoulos, N.; Eton, O.; Ellerhorst, J.; Smith, T. Phase II evaluation of bryostatin-1 in metastatic melanoma. Melanoma Res. 2001, 11, 183–188. [Google Scholar] [CrossRef]
- Varterasian, M.L.; Pemberton, P.A.; Hulburd, K.; Rodriguez, D.H.; Murgo, A.; Al-Katib, A.M. Phase II study of bryostatin 1 in patients with relapsed multiple myeloma. Invest. New Drugs 2001, 19, 245–247. [Google Scholar] [CrossRef]
- Brockstein, B.; Samuels, B.; Humerickhouse, R.; Arietta, R.; Fishkin, P.; Wade, J.; Sosman, J.; Vokes, E.E. Phase II studies of bryostatin-1 in patients with advanced sarcoma and advanced head and neck cancer. Invest. New Drugs 2001, 19, 249–254. [Google Scholar] [CrossRef]
- Pfister, D.G.; McCaffrey, J.; Zahalsky, A.J.; Schwartz, G.K.; Lis, E.; Gerald, W.; Huvos, A.; Shah, J.; Kraus, D.; Shaha, A.; et al. A phase II trial of bryostatin-1 in patients with metastatic or recurrent squamous cell carcinoma of the head and neck. Invest. New Drugs 2002, 20, 123–127. [Google Scholar] [CrossRef]
- Tozer, R.G.; Burdette-Radoux, S.; Berlanger, K.; Davis, M.L.; Lohmann, R.C.; Rusthoven, J.R.; Wainman, N.; Zee, B.; Seymour, L. A randomized phase II study of two schedules of bryostatin-1 (NSC339555) in patients with advanced malignant melanoma: A National Cancer Institute of Canada Clinical Trials Group study. Invest. New Drugs 2002, 20, 407–412. [Google Scholar] [CrossRef]
- Haas, N.B.; Smith, M.; Lewis, N.; Littman, L.; Yeslow, G.; Joshi, I.D.; Murgo, A.; Bradley, J.; Gordon, R.; Wang, H.; et al. Weekly bryostatin-1 in metastatic renal cell carcinoma: A phase II study. Clin. Cancer Res. 2003, 9, 109–114. [Google Scholar]
- Winegarden, J.D.; Mauer, A.M.; Gajewski, T.F.; Hoffman, P.C.; Krauss, S.; Rudin, C.M.; Vokes, E.E. A phase II study of bryostatin-1 and paclitaxel in patients with advanced non-small cell lung cancer. Lung Cancer 2003, 39, 191–196. [Google Scholar] [CrossRef]
- Clamp, A.R.; Blackhall, F.H.; Vasey, P.; Soukop, M.; Coleman, R.; Halbert, G.; Robson, L.; Jayson, G.C. A phase II trial of bryostatin-1 administered by weekly 24-hour infusion in recurrent epithelial ovarian carcinoma. Br. J. Cancer 2003, 89, 1152–1154. [Google Scholar]
- Madhusudan, S.; Protheroe, A.; Propper, D.; Han, C.; Corrie, P.; Earl, H.; Hancock, B.; Vasey, P.; Turner, A.; Balkwill, F.; et al. A multicentre phase II trial of bryostatin-1 in patients with advanced renal cancer. Br. J. Cancer 2003, 89, 1418–1422. [Google Scholar]
- Armstrong, D.K.; Blessing, J.A.; Look, K.Y.; Schilder, R.; Nunez, E.R. A randomized phase II evaluation of bryostatin-1 (NSC #339555) in recurrent or persistent platinum-sensitive ovarian cancer: A Gynecologic Oncology Group Study. Invest. New Drugs 2003, 21, 373–377. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Blessing, J.A.; Rader, J.; Sorosky, J.I. A randomized phase II evaluation of bryostatin-1 (NSC #339555) in persistent or recurrent squamous cell carcinoma of the cervix: A Gynecologic Oncology Group Study. Invest. New Drugs 2003, 21, 453–457. [Google Scholar] [CrossRef]
- Peterson, A.C.; Harlin, H.; Karrison, T.; Vogelzang, N.J.; Knost, J.A.; Kugler, J.W.; Lester, E.; Vokes, E.; Gajewski, T.F.; Stadler, W.M. A randomized phase II trial of interleukin-2 in combination with four different doses of bryostatin-1 in patients with renal cell carcinoma. Invest. New Drugs 2006, 24, 141–149. [Google Scholar] [CrossRef]
- Ajani, J.A.; Jiang, Y.; Faust, J.; Chang, B.B.; Ho, L.; Yao, J.C.; Rousey, S.; Dakhil, S.; Cherny, R.C.; Craig, C.; Bleyer, A. A multi-center phase II study of sequential paclitaxel and bryostatin-1 (NSC 339555) in patients with untreated, advanced gastric or gastroesophageal junction adenocarcinoma. Invest. New Drugs 2006, 24, 353–357. [Google Scholar] [CrossRef]
- Ku, G.Y.; Ilson, D.H.; Schwartz, L.H.; Capanu, M.; O’Reilly, E.; Shah, M.A.; Kelsen, D.P.; Schwartz, G.K. Phase II trial of sequential paclitaxel and 1 h infusion of bryostatin-1 in patients with advanced esophageal cancer. Cancer Chemother. Pharmacol. 2008, 62, 875–880. [Google Scholar] [CrossRef]
- Barr, P.M.; Lazarus, H.M.; Cooper, B.W.; Schluchter, M.D.; Panneerselvam, A.; Jacobberger, J.W.; Hsu, J.W.; Janakiraman, N.; Simic, A.; Dowlati, A.; et al. Phase II study of bryostatin 1 and vincristine for aggressive non-Hodgkin lymphoma relapsing after an autologous stem cell transplant. Am. J. Hematol. 2009, 84, 484–487. [Google Scholar] [CrossRef]
- Lam, A.P.; Sparano, J.A.; Vinciguerra, V.; Ocean, A.J.; Christos, P.; Hochster, H.; Camacho, F.; Goel, S.; Mani, S.; Kaubisch, A. Phase II study of paclitaxel plus the protein kinase C inhibitor bryostatin-1 in advanced pancreatic carcinoma. Am. J. Clin. Oncol. 2010, 33, 121–124. [Google Scholar]
- Morgan, R.J., Jr.; Leong, L.; Chow, W.; Gandara, D.; Frankel, P.; Garcia, A.; Lenz, H.J.; Doroshow, J.H. Phase II trial of bryostatin-1 in combination with cisplatin in patients with recurrent or persistent epithelial ovarian cancer: A California cancer consortium study. Invest. New Drugs 2012, 30, 723–728. [Google Scholar] [CrossRef]
- Clamp, A.; Jayson, G.C. The clinical development of the bryostatins. Anticancer Drugs 2002, 13, 673–683. [Google Scholar] [CrossRef]
- Hale, K.J.; Hummersone, M.G.; Manaviazar, S.; Frigerio, M. The chemistry and biology of the bryostatin antitumour macrolides. Nat. Prod. Rep. 2002, 19, 413–453. [Google Scholar] [CrossRef]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: http://clinicaltrials.gov/ct2/results?term=panobinostat&Search=Search (accessed on 1 February 2013).
- Hainsworth, J.D.; Infante, J.R.; Spigel, D.R.; Arrowsmith, E.R.; Boccia, R.V.; Burris, H.A. A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Invest. 2011, 29, 451–455. [Google Scholar]
- Dimicoli, S.; Jabbour, E.; Borthakur, G.; Kadia, T.; Estrov, Z.; Yang, H.; Kelly, M.; Pierce, S.; Kantarjian, H.; Garcia-Manero, G. Phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in patients with low or intermediate-1 risk myelodysplastic syndrome. Am. J. Hematol. 2012, 87, 127–129. [Google Scholar]
- Wolf, J.L.; Siegel, D.; Goldschmidt, H.; Hazell, K.; Bourquelot, P.M.; Bengoudifa, B.R.; Matous, J.; Vij, R.; de Magalhaes-Silverman, M.; Abonour, R.; et al. Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leuk. Lymphoma 2012, 53, 1820–1823. [Google Scholar] [CrossRef]
- Offidani, M.; Polloni, C.; Cavallo, F.; Liberati, A.M.; Ballanti, S.; Pulini, S.; Catarini, M.; Alesiani, F.; Corvatta, L.; Gentili, S.; et al. Phase II study of melphalan, thalidomide and prednisone combined with oral panobinostat in patients with relapsed/refractory multiple myeloma. Leuk. Lymphoma 2012, 53, 1722–1727. [Google Scholar] [CrossRef]
- Wang, H.B.; Cao, Q.; Dudek, A.Z. Phase II study of panobinostat and bortezomib in patients with pancreatic cancer progressing on gemcitabine-based therapy. Anticancer Res. 2012, 32, 1027–1031. [Google Scholar]
- Younes, A.; Sureda, A.; Ben-Yehuda, D.; Zinzani, P.L.; Ong, T.C.; Prince, H.M.; Harrison, S.J.; Kirschbaum, M.; Johnston, P.; Gallagher, J.; et al. Panobinostat in patients with relapsed/refractory Hodgkin’s Lymphoma after autologous stem-cell transplantation: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2197–2203. [Google Scholar] [CrossRef]
- Duvic, M.; Dummer, R.; Becker, J.C.; Poulalhon, N.; Ortiz Romero, P.; Grazia Bernengo, M.; Lebbe, C.; Assaf, C.; Squier, M.; Williams, D.; et al. Panobinostat activity in both bexarotene-exposed and -naive patients with refractory cutaneous T-cell lymphoma: Results of a phase II trial. Eur. J. Cancer 2013, 49, 386–394. [Google Scholar]
- Ghobrial, I.M.; Campigotto, F.; Murphy, T.J.; Boswell, E.N.; Banwait, R.; Azab, F.; Chuma, S.; Kunsman, J.; Donovan, A.; Masood, F.; et al. Results of the phase II trial of single agent histone deacetylase inhibitor panobinostat in patients with relapsed/refractory Waldenstrom macroglobulinemia. Blood 2013, 121, 1296–1303. [Google Scholar]
- Jones, S.F.; Infante, J.R.; Thompson, D.S.; Mohyuddin, A.; Bendell, J.C.; Yardley, D.A.; Burris, H.A., III. A phase I trial of oral administration of panobinostat in combination with paclitaxel and carboplatin in patients with solid tumors. Cancer Chemother. Pharmacol. 2012, 70, 471–475. [Google Scholar] [CrossRef]
- Strickler, J.H.; Starodub, A.N.; Jia, J.; Meadows, K.L.; Nixon, A.B.; Dellinger, A.; Morse, M.A.; Uronis, H.E.; Marcom, P.K.; Zafar, S.Y.; et al. Phase I study of bevacizumab, everolimus, and panobinostat (LBH-589) in advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 70, 251–258. [Google Scholar] [CrossRef]
- Prince, H.M.; Bishton, M.J.; Harrison, S.J. Clinical studies of histone deacetylase inhibitors. Clin. Cancer Res. 2009, 15, 3958–3969. [Google Scholar] [CrossRef]
- Barboza, N.M.; Medina, D.J.; Budak-Alpdogan, T.; Aracil, M.; Jimeno, J.M.; Bertino, J.R.; Banerjee, D. Plitidepsin (Aplidin) is a potent inhibitor of diffuse large cell and Burkitt lymphoma and is synergistic with rituximab. Cancer Biol. Ther. 2012, 13, 114–122. [Google Scholar] [CrossRef]
- Soto-Matos, A.; Szyldergemajn, S.; Extremera, S.; Miguel-Lillo, B.; Alfaro, V.; Coronado, C.; Lardelli, P.; Roy, E.; Corrado, C.S.; Kahatt, C. Plitidepsin has a safe cardiac profile: A comprehensive analysis. Mar. Drugs 2011, 9, 1007–1023. [Google Scholar] [CrossRef]
- Geoerger, B.; Estlin, E.J.; Aerts, I.; Kearns, P.; Gibson, B.; Corradini, N.; Doz, F.; Lardelli, P.; Miguel, B.D.; Soto, A.; et al. A phase I and pharmacokinetic study of plitidepsin in children with advanced solid tumours: An Innovative Therapies for Children with Cancer (ITCC) study. Eur. J. Cancer 2012, 48, 289–296. [Google Scholar] [CrossRef]
- Schoffski, P.; Guillem, V.; Garcia, M.; Rivera, F.; Tabernero, J.; Cullell, M.; Lopez-Martin, J.A.; Pollard, P.; Dumez, H.; del Muro, X.G.; et al. Phase II randomized study of Plitidepsin (Aplidin), alone or in association with l-carnitine, in patients with unresectable advanced renal cell carcinoma. Mar. Drugs 2009, 7, 57–70. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Raymond, E.; Faivre, S. Aplidine: A paradigm of how to handle the activity and toxicity of a novel marine anticancer poison. Curr. Pharm. Des. 2007, 13, 3427–3439. [Google Scholar] [CrossRef]
- Jimeno, J.; Lopez-Martin, J.A.; Ruiz-Casado, A.; Izquierdo, M.A.; Scheuer, P.J.; Rinehart, K. Progress in the clinical development of new marine-derived anticancer compounds. Anticancer Drugs 2004, 15, 321–329. [Google Scholar] [CrossRef]
- Baudin, E.; Droz, J.P.; Paz-Ares, L.; van Oosterom, A.T.; Cullell-Young, M.; Schlumberger, M. Phase II study of plitidepsin 3-hour infusion every 2 weeks in patients with unresectable advanced medullary thyroid carcinoma. Am. J. Clin. Oncol. 2010, 33, 83–88. [Google Scholar] [CrossRef]
- Eisen, T.; Thomas, J.; Miller, W.H.; Gore, M.; Wolter, P.; Kavan, P.; Martin, J.A.L.; Lardelli, P. Phase II study of biweekly plitidepsin as second-line therapy in patients with advanced malignant melanoma. Melanoma Res. 2009, 19, 185–192. [Google Scholar] [CrossRef]
- Eisen, T.; Thatcher, N.; Leyvraz, S.; Miller, W.H.; Couture, F.; Lorigan, P.; Luthi, F.; Small, D.; Tanovic, A.; O’Brien, M. Phase II study of weekly plitidepsin as second-line therapy for small cell lung cancer. Lung Cancer 2009, 64, 60–65. [Google Scholar] [CrossRef]
- Peschel, C.; Hartmann, J.T.; Schmittel, A.; Bokemeyer, C.; Schneller, F.; Keilholz, U.; Buchheidt, D.; Millan, S.; Izquierdo, M.A.; Hofheinz, R.D. Phase II study of plitidepsin in pretreated patients with locally advanced or metastatic non-small cell lung cancer. Lung Cancer 2008, 60, 374–380. [Google Scholar] [CrossRef]
- Ribrag, V.; Caballero, D.; Ferme, C.; Zucca, E.; Arranz, R.; Briones, J.; Gisselbrecht, C.; Salles, G.; Gianni, A.M.; Gomez, H.; et al. Multicenter phase II study of plitidepsin in patients with relapsed/refractory non-Hodgkin’s lymphoma. Haematologica 2012, 98, 357–363. [Google Scholar]
- Mateos, M.V.; Cibeira, M.T.; Richardson, P.G.; Prosper, F.; Oriol, A.; de la Rubia, J.; Lahuerta, J.J.; Garcia-Sanz, R.; Extremera, S.; Szyldergemajn, S.; et al. Phase II clinical and pharmacokinetic study of plitidepsin 3-hour infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clin. Cancer Res. 2010, 16, 3260–3269. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Hideshima, T.; Chauhan, D.; McMillin, D.W.; Klippel, S.; Laubach, J.P.; Munshi, N.C.; Anderson, K.C.; Richardson, P.G. Emerging treatments for multiple myeloma: Beyond immunomodulatory drugs and bortezomib. Semin. Hematol. 2009, 46, 166–175. [Google Scholar] [CrossRef]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest. New Drugs 2012, 30, 2303–2317. [Google Scholar] [CrossRef]
- Hamlin, P.A.; Aghajanian, C.; Younes, A.; Hong, D.S.; Palladino, M.A.; Longenecker, A.M.; Lloyd, G.K.; Hannah, A.L.; Spear, M.A.; Kurzrock, R. First-in-human phase I study of the novel structure proteasome inhibitor NPI-0052. J. Clin. Oncol. 2009, 27, 3516. [Google Scholar]
- Hofmeister, C.C.; Richardson, P.; Zimmerman, T.; Spear, M.A.; Palladino, M.A.; Longenecker, A.M.; Cropp, G.F.; Lloyd, G.K.; Hannah, A.L.; Anderson, K. Clinical trial of the novel structure proteasome inhibitor NPI-0052 in patients with relapsed and relapsed/refractory multiple myeloma (r/r MM). J. Clin. Oncol. 2009, 27, 8505. [Google Scholar]
- Richardson, P.; Hofmeister, C.C.; Zimmerman, T.M.; Chanan-Khan, A.A.; Spear, M.A.; Palladino, M.A.; Longenecker, A.M.; Cropp, G.; Lloyd, G.K.; Wear, S.; et al. Phase 1 clinical trial of NPI-0052, a novel proteasome inhibitor in patients with multiple myeloma. Blood 2008, 112, 955–956. [Google Scholar]
- Lawasut, P.; Chauhan, D.; Laubach, J.; Hayes, C.; Fabre, C.; Maglio, M.; Mitsiades, C.; Hideshima, T.; Anderson, K.C.; Richardson, P.G. New proteasome inhibitors in myeloma. Curr. Hematol. Malig. Rep. 2012, 7, 258–266. [Google Scholar] [CrossRef]
- Mita, M.M.; Spear, M.A.; Yee, L.K.; Mita, A.C.; Heath, E.I.; Papadopoulos, K.P.; Federico, K.C.; Reich, S.D.; Romero, O.; Malburg, L.; et al. Phase 1 first-in-human trial of the vascular disrupting agent plinabulin(NPI-2358) in patients with solid tumors or lymphomas. Clin. Cancer Res. 2010, 16, 5892–5899. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, Y.-Q.; Miao, Z.-H. Marine-Derived Angiogenesis Inhibitors for Cancer Therapy. Mar. Drugs 2013, 11, 903-933. https://doi.org/10.3390/md11030903
Wang Y-Q, Miao Z-H. Marine-Derived Angiogenesis Inhibitors for Cancer Therapy. Marine Drugs. 2013; 11(3):903-933. https://doi.org/10.3390/md11030903
Chicago/Turabian StyleWang, Ying-Qing, and Ze-Hong Miao. 2013. "Marine-Derived Angiogenesis Inhibitors for Cancer Therapy" Marine Drugs 11, no. 3: 903-933. https://doi.org/10.3390/md11030903
APA StyleWang, Y. -Q., & Miao, Z. -H. (2013). Marine-Derived Angiogenesis Inhibitors for Cancer Therapy. Marine Drugs, 11(3), 903-933. https://doi.org/10.3390/md11030903