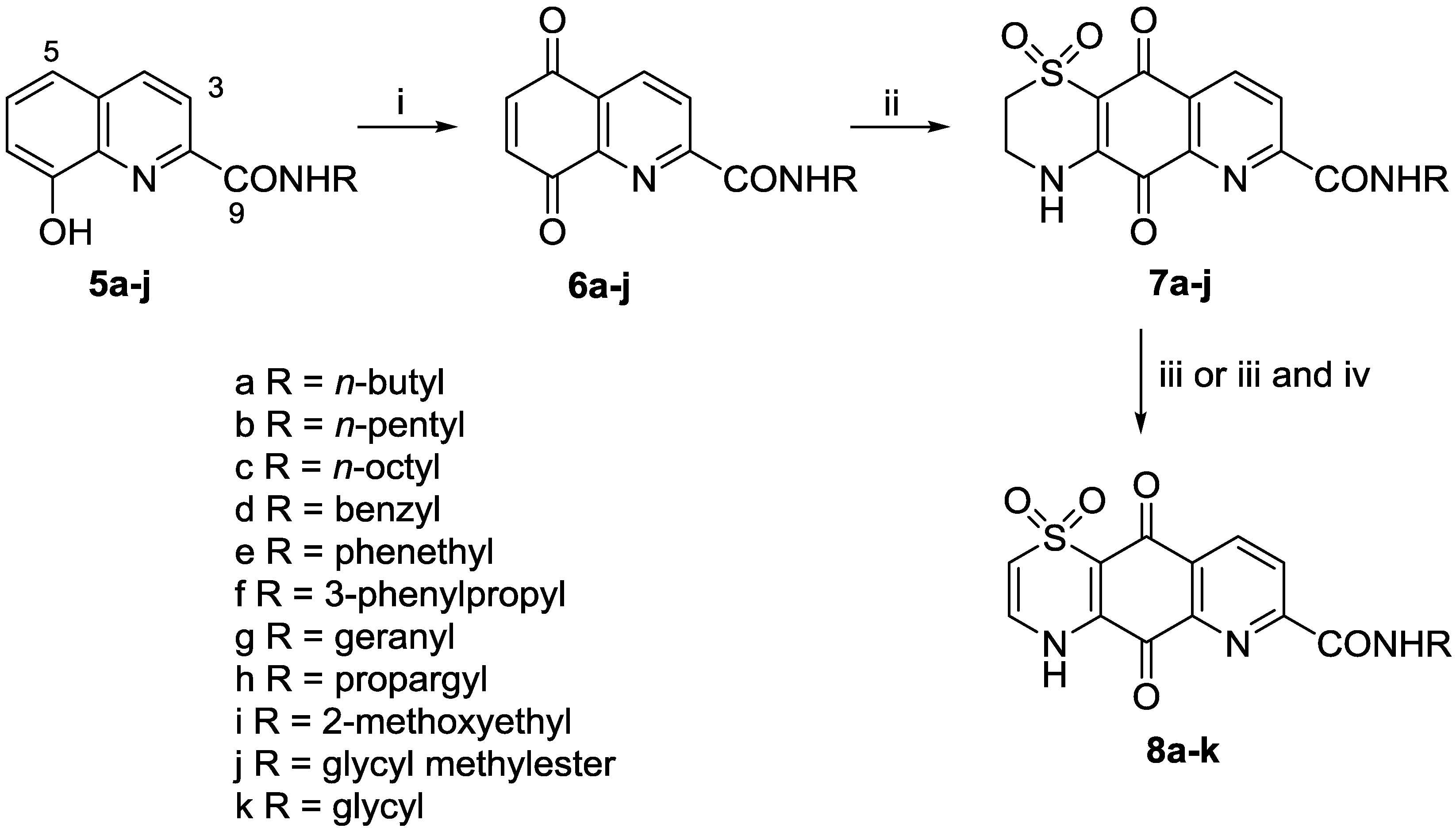
3.2.1. General Procedure for the Preparation of 8-Hydroxyquinoline-2-carboxamides 5a–5j
To a solution of 8-hydroxyquinoline-2-carboxylic acid and PyBOP (1.25 equiv.) in dry DMF (3–6 mL), amine (1–2 equiv.) and triethylamine (1.25 equiv.) were added under N2. The reaction mixture was then stirred under N2 at rt for 12 h, after which time the mixture was dried in vacuo. The residue was purified by reversed-phase C18 flash column chromatography (0%–80% MeOH in H2O (0.05% TFA)) and silica gel column chromatography (0%–1% MeOH in CH2Cl2).
3.2.1.1. N-n-Butyl-8-hydroxyquinoline-2-carboxamide (5a)
From 8-hydroxyquinoline-2-carboxylic acid (100 mg, 0.529 mmol), PyBOP (330 mg, 0.64 mmol), n-butylamine (104 µL, 1.05 mmol) and triethylamine (88 µL, 0.632 mmol) in DMF (6 mL) to give 5a as a yellow oil (108 mg, 84% yield).
Rf = 0.68 (1% MeOH/CH2Cl2); IR νmax (ATR) 3291, 1650, 1539, 1502, 1465, 1159 cm−1; 1H NMR (CDCl3, 400 MHz) δH 8.34 (1H, d, J = 8.4 Hz, H-3), 8.29 (1H, d, J = 8.4 Hz, H-4), 8.01 (1H, br s, NH-2′), 7.86 (1H, br s, OH), 7.53 (1H, t, J = 7.8 Hz, H-6), 7.40 (1H, d, J = 8.4 Hz, H-5), 7.23 (1H, d, J = 8.0 Hz, H-7), 3.53 (2H, dt, J = 7.2, 7.2 Hz, H2-3′), 1.63 (2H, p, J = 7.3 Hz, H2-4′), 1.40 (2H, sex., J = 7.6 Hz, H2-5′), 0.92 (3H, t, J = 7.6 Hz, H3-6′); 13C NMR (CDCl3, 100 MHz) δC 164.1 (C-1′), 152.2 (C-8), 148.2 (C-2), 137.8 (C-4), 136.1 (C-8a), 129.7 (C-4a), 129.2 (C-6), 119.9 (C-3), 118.3 (C-5), 111.2 (C-7), 39.5 (C-3′), 31.8 (C-4′), 20.2 (C-5′), 13.8 (C-6′); (+)-ESIMS m/z 245 [M + H]+; (+)-HRESIMS m/z 245.1287 [M + H]+ (calcd. for C14H17N2O2, 245.1285).
3.2.1.2. N-n-Pentyl-8-hydroxyquinoline-2-carboxamide (5b)
From 8-hydroxyquinoline-2-carboxylic acid (50 mg, 0.26 mmol), PyBOP (165 mg, 0.32 mmol), n-pentylamine (61 µL, 0.53 mmol) and triethylamine (44 µL, 0.32 mmol) in DMF (3 mL) to give 5b as a colorless oil (65 mg, 97% yield).
Rf = 0.65 (5% MeOH/CH2Cl2); IR νmax (ATR) 3266, 2929, 1647, 1500 cm−1; 1H NMR (CDCl3, 400 MHz) δH 8.46 (1H, t, J = 5.8 Hz, NH-2′), 8.33 (1H, d, J = 8.6 Hz, H-3), 8.24 (1H, d, J = 8.6 Hz, H-4), 7.49 (1H, t, J = 8.2 Hz, H-6), 7.34 (1H, d, J = 8.2 Hz, H-5), 7.20 (1H, d, J = 8.2 Hz, H-7), 3.46 (2H, dt, J = 7.4, 5.8 Hz, H2-3′), 1.58 (2H, p, J = 7.4 Hz, H2-4′), 1.30–1.18 (4H, m, H2-5′/H2-6′), 0.81 (3H, t, J = 7.4 Hz, H3-7′); 13C NMR (CDCl3, 100 MHz) δC 164.4 (C-1′), 152.4 (C-8), 148.0 (C-2), 137.6 (C-4), 136.6 (C-8a), 129.7 (C-4a), 129.2 (C-6), 119.7 (C-3), 118.1 (C-5), 111.2 (C-7), 39.8 (C-3′), 29.4 (C-4′), 29.1 (C-5′), 22.3 (C-6′), 13.8 (C-7′); (+)-ESIMS m/z 281 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 281.1259 (calcd. for C15H18N2NaO2, 281.1260).
3.2.1.3. N-n-Octyl-8-hydroxyquinoline-2-carboxamide (5c)
From 8-hydroxyquinoline-2-carboxylic acid (50 mg, 0.26 mmol), PyBOP (165 mg, 0.32 mmol), n-octylamine (87 µL, 0.527 mmol) and triethylamine (44 µL, 0.32 mmol) in DMF (3 mL) to give 5c as a colorless oil (73 mg, 94% yield).
Rf = 0.80 (5% MeOH/CH2Cl2); IR νmax (ATR) 3297, 2924, 1648, 1501 cm−1; 1H NMR (CDCl3, 400 MHz) δH 8.69 (1H, t, J = 5.7 Hz, NH-2′), 8.33 (1H, d, J = 8.6 Hz, H-3), 8.22 (1H, d, J = 8.6 Hz, H-4), 7.48 (1H, t, J = 8.0 Hz, H-6), 7.32 (1H, d, J = 8.0 Hz, H-5), 7.18 (1H, d, J = 8.0 Hz, H-7), 3.45 (2H, dt, J = 7.2, 5.7 Hz, H2-3′), 1.55 (2H, p, J = 7.2 Hz, H2-4′), 1.27–1.09 (10H, m, H2-5′/H2-6′/H2-7′/H2-8′/H2-9′), 0.80 (3H, t, J = 7.2 Hz, H3-10′); 13C NMR (CDCl3, 100 MHz) δC 164.6 (C-1′), 152.5 (C-8), 147.8 (C-2), 137.6 (C-4), 136.6 (C-8a), 129.6 (C-4a), 129.2 (C-6), 119.6 (C-3), 118.1 (C-5), 111.2 (C-7), 39.9 (C-3′), 31.7 (C-6′*), 29.6 (C-4′), 29.2 (C-7′*), 29.1 (C-8′*), 27.0 (C-5′), 22.5 (C-9′*), 14.0 (C-10′); (+)-ESIMS m/z 323 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 323.1740 (calcd. for C18H24N2NaO2, 323.1730).
3.2.1.4. N-Benzyl-8-hydroxyquinoline-2-carboxamide (5d)
From 8-hydroxyquinoline-2-carboxylic acid (50 mg, 0.26 mmol), PyBOP (165 mg, 0.32 mmol), benzylamine (58 µL, 0.53 mmol) and triethylamine (44 µL, 0.32 mmol) in DMF (3 mL) to give 5d as a colorless oil (57 mg, 79% yield).
Rf = 0.72 (5% MeOH/CH2Cl2); IR νmax (ATR) 3251, 3062, 1642, 1501 cm−1; 1H NMR (CDCl3, 400 MHz) δH 8.90 (1H, br s, NH-2′), 8.29 (1H, d, J = 8.4 Hz, H-3), 8.20 (1H, d, J = 8.4 Hz, H-4), 7.48 (1H, t, J = 7.9 Hz, H-6), 7.33 (1H, d, J = 7.9 Hz, H-5), 7.25–7.13 (6H, m, H-7/2H-5′/2H-6′/H-7′), 4.61 (2H, br s, H2-3′); 13C NMR (CDCl3, 100 MHz) δC 164.6 (C-1′), 152.4 (C-8), 147.5 (C-2), 137.9 (C-4′), 137.6 (C-4), 136.5 (C-8a), 129.7 (C-4a), 129.3 (C-6), 128.5 (C-5′), 127.7 (C-6′), 127.3 (C-7′), 119.7 (C-3), 118.1 (C-5), 111.3 (C-7), 43.6 (C-3′); (+)-ESIMS m/z 301 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 301.0949 (calcd. for C17H14N2NaO2, 301.0947).
3.2.1.5. N-Phenethyl-8-hydroxyquinoline-2-carboxamide (5e)
From 8-hydroxyquinoline-2-carboxylic acid (50 mg, 0.26 mmol), PyBOP (165 mg, 0.32 mmol), phenethylamine (66 µL, 0.53 mmol) and triethylamine (44 µL, 0.32 mmol) in DMF (3 mL) to give 5e as a colorless oil (65 mg, 86% yield).
Rf = 0.65 (5% MeOH/CH2Cl2); IR νmax (ATR) 3288, 3073, 1643, 1501 cm−1; 1H NMR (CDCl3, 300 MHz) δH 8.33 (1H, d, J = 8.5 Hz, H-3), 8.28 (1H, d, J = 8.5 Hz, H-4), 7.53 (1H, t, J = 7.8 Hz, H-6), 7.40–7.20 (7H, m, H-5/H-7/2H-6′/2H-7′/H-8′), 3.77 (2H, dt, J = 7.0, 6.8 Hz, H2-3′), 2.96 (2H, t, J = 7.0 Hz, H2-4′); 13C NMR (CDCl3, 75 MHz) δC 164.1 (C-1′), 152.3 (C-8), 147.9 (C-2), 138.8 (C-5′), 137.8 (C-4), 136.5 (C-8a), 129.7 (C-4a), 129.3 (C-6), 128.8 (C-6′), 128.7 (C-7′), 126.7 (C-8′), 119.7 (C-3), 118.1 (C-5), 111.2 (C-7), 40.7 (C-3′), 35.8 (C-4′); (+)-ESIMS m/z 293 [M + H]+; (+)-HRESIMS m/z [M + H]+ 293.1292 (calcd. for C18H17N2O2, 293.1285).
3.2.1.6. N-(3-Phenylpropyl)-8-hydroxyquinoline-2-carboxamide (5f)
From 8-hydroxyquinoline-2-carboxylic acid (50 mg, 0.26 mmol), PyBOP (165 mg, 0.32 mmol), 3-phenylpropylamine (66 µL, 0.46 mmol) and triethylamine (44 µL, 0.32 mmol) in DMF (3 mL) to give 5f as a colorless oil (67 mg, 84% yield).
Rf = 0.65 (5% MeOH/CH2Cl2); IR νmax (ATR) 3257, 2929, 1647, 1500 cm−1; 1H NMR (CDCl3, 400 MHz) δH 8.64 (1H, br s, NH-2′), 8.32 (1H, d, J = 8.5 Hz, H-3), 8.20 (1H, d, J = 8.5 Hz, H-4), 7.49 (1H, t, J = 7.9 Hz, H-6), 7.33 (1H, d, J = 7.9 Hz, H-5), 7.22–7.00 (6H, m, H-7/2H-7′/2H-8′/H-9′), 3.53–3.46 (2H, m, H2-3′), 2.57 (2H, t, J = 7.2 Hz, H2-5′), 1.89 (2H, p, J = 7.2 Hz, H2-4′); 13C NMR (CDCl3, 100 MHz) δC 164.6 (C-1′), 152.5 (C-8), 147.7 (C-2), 141.2 (C-6′), 137.6 (C-4), 136.5 (C-8a), 129.6 (C-4a), 129.2 (C-6), 128.2 (C-7′), 128.1 (C-8′), 125.8 (C-9′), 119.5 (C-3), 118.1 (C-5), 111.2 (C-7), 39.4 (C-3′), 33.2 (C-5′), 31.0 (C-4′); (+)-ESIMS m/z 329 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 329.1267 (calcd. for C19H18N2NaO2, 329.1260).
3.2.1.7. N-Geranyl-8-hydroxyquinoline-2-carboxamide (5g)
From 8-hydroxyquinoline-2-carboxylic acid (50 mg, 0.26 mmol), PyBOP (165 mg, 0.32 mmol), geranylamine (98 µL, 0.53 mmol) and triethylamine (44 µL, 0.32 mmol) in DMF (3 mL) to give 5g as a colorless oil (85 mg, 100% yield).
Rf = 0.66 (5% MeOH/CH2Cl2); IR νmax (ATR) 3276, 2914, 1646, 1501 cm−1; 1H NMR (CDCl3, 300 MHz) δH 8.38 (1H, t, J = 5.6 Hz, NH-2′), 8.33 (1H, d, J = 8.5 Hz, H-3), 8.23 (1H, d, J = 8.5 Hz, H-4), 7.49 (1H, t, J = 6.1 Hz, H-6), 7.34 (1H, dd, J = 6.1, 1.1 Hz, H-5), 7.19 (1H, dd, J = 6.1, 1.1 Hz, H-7), 5.27 (1H, t, J = 6.9 Hz, H-4′), 4.99 (1H, t, J = 6.9 Hz, H-8′), 4.12 (2H, dd, J = 6.3, 6.3 Hz, H2-3′), 2.04–1.95 (2H, m, H2-7′), 1.93–1.86 (2H, m, H2-6′), 1.62 (6H, s, H3-11′/H3-12′), 1.53 (3H, s, H3-10′); 13C NMR (CDCl3, 75 MHz) δC 164.2 (C-1′), 152.4 (C-8), 148.0 (C-2), 139.7 (C-5′), 137.6 (C-4), 136.5 (C-8a), 131.6 (C-9′), 129.6 (C-4a), 129.2 (C-6), 123.7 (C-8′), 119.7 (C-4′), 119.6 (C-3), 118.1 (C-5), 111.2 (C-7), 39.4 (C-6′), 37.7 (C-3′), 26.3 (C-7′), 25.6 (C-12′), 17.6 (C-10′), 16.3 (C-11′); (+)-ESIMS m/z 325 [M + H]+; (+)-HRESIMS m/z [M + H]+ 325.1903 (calcd. for C20H25N2O2, 325.1911).
3.2.1.8. N-Propargyl-8-hydroxyquinoline-2-carboxamide (5h)
From 8-hydroxyquinoline-2-carboxylic acid (50 mg, 0.26 mmol), PyBOP (165 mg, 0.32 mmol), propargylamine (29 mg, 0.53 mmol) and triethylamine (44 µL, 0.32 mmol) in DMF (3mL) to give 5h as a colorless oil (47 mg, 80% yield).
Rf = 0.56 (10% MeOH/CH2Cl2); IR νmax (ATR) 3303, 3273, 1646, 1504 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 10.17 (1H, br s, OH-9), 9.99 (1H, t, J = 5.8 Hz, NH-2′), 8.49 (1H, d, J = 8.4 Hz, H-4), 8.14 (1H, d, J = 8.4 Hz, H-3), 7.56 (1H, t, J = 7.8 Hz, H-6), 7.46 (1H, d, J = 7.8 Hz, H-5), 7.18 (1H, d, J = 7.8, Hz, H-7), 4.23 (2H, dd, J = 5.8, 2.4 Hz, H2-3′), 3.22 (1H, t, J = 2.4 Hz, H2-5′); 13C NMR (DMSO-d6, 100 MHz) δC 163.5 (C-1′), 153.7 (C-8), 147.0 (C-2), 137.8 (C-4), 136.4 (C-8a), 129.6 (C-4a and C-6), 118.8 (C-3), 117.5 (C-5), 111.6 (C-7), 81.1 (C-4′), 73.3 (C-5′), 28.1 (C-3′); (+)-ESIMS m/z 227 [M + H]+; (+)-HRESIMS m/z [M + H]+ 227.0814 (calcd. for C13H11N2O2, 227.0815).
3.2.1.9. N-(2-Methoxyethyl)-8-hydroxyquinoline-2-carboxamide (5i)
From 8-hydroxyquinoline-2-carboxylic acid (100 mg, 0.529 mmol), PyBOP (330 mg, 0.64 mmol), 2-methoxyethylamine (92.6 µL, 1.07 mmol) and triethylamine (88 µL, 0.632 mmol) in DMF (6 mL) to give 5i as a colorless oil (106 mg, 82% yield).
Rf = 0.83 (10% MeOH/CH2Cl2); IR νmax (ATR) 3315, 1648, 1542, 1501, 1120 cm−1; 1H NMR (CDCl3, 400 MHz) δH 8.68 (1H, br s, NH-2′), 8.38 (1H, br s, OH), 8.32 (1H, d, J = 8.4 Hz, H-3), 8.26 (1H, d, J = 8.4 Hz, H-4), 7.50 (1H, t, J = 8.0 Hz, H-6), 7.36 (1H, d, J = 8.4 Hz, H-5), 7.21 (1H, d, J = 7.6 Hz, H-7), 3.73 (2H, dt, J = 5.6, 5.2 Hz, H2-3′), 3.61 (2H, t, J = 5.2 Hz, H2-4′), 3.36 (3H, s, H3-5′); 13C NMR (CDCl3, 100 MHz) δC 164.6 (C-1′), 152.6 (C-8), 147.9 (C-2), 137.6 (C-4), 136.6 (C-8a), 129.7 (C-4a), 129.3 (C-6), 119.7 (C-3), 118.1 (C-5), 111.3 (C-7), 71.3 (C-4′), 58.8 (C-5′), 39.5 (C-3′); (+)-ESIMS m/z 247 [M + H]+; (+)-HRESIMS m/z 247.1076 [M + H]+ (calcd. for C13H15N2O3, 247.1077).
3.2.1.10. N-Glycine(methylester)-8-hydroxyquinoline-2-carboxamide (5j)
From 8-hydroxyquinoline-2-carboxylic acid (100 mg, 0.529 mmol), PyBOP (330 mg, 0.64 mmol), glycine methyl ester hydrochloride (94.4 mg, 0.76 mmol) and triethylamine (220 µL, 1.58 mmol) in DMF (6 mL) to give 5j as a colorless oil (128 mg, 93% yield).
Rf = 0.86 (10% MeOH/CH2Cl2); 1H NMR (CDCl3, 400 MHz) δH 9.08 (1H, t, J = 6.1 Hz, NH-2′), 8.38 (1H, br s, OH), 8.03 (2H, s, H-3 and H-4), 7.43 (1H, t, J = 8.0 Hz, H-6), 7.23 (1H, dd, J = 8.4, 1.0 Hz, H-5), 7.13 (1H, dd, J = 7.6, 1.0 Hz, H-7), 4.27 (2H, d, J = 6.1 Hz, H2-3′), 3.72 (3H, s, H3-5′); 13C NMR (CDCl3, 100 MHz) δC 171.2 (C-4′), 165.0 (C-1′), 152.7 (C-8), 146.8 (C-2), 137.2 (C-4), 136.4 (C-8a), 129.6 (C-4a), 129.5 (C-6), 119.3 (C-3), 117.9 (C-5), 111.3 (C-7), 52.5 (C-5′), 41.3 (C-3′); (+)-ESIMS m/z 261 [M + H]+; (+)-HRESIMS m/z 261.0863 [M + H]+ (calcd. for C13H13N2O4, 261.0870).
3.2.2. General Procedure for Preparation of Quinones 6a–6j
A solution of PIFA (2–3 equiv.) in MeCN/H2O (2:1 mL) was cooled to 0 °C, followed by the addition of the appropriate 8-hydroxyquinoline-2-carboxamide in CH2Cl2 (1 mL). The dark brown suspension was stirred for 20 min. at 0 °C before being poured into a mixture of CH2Cl2 (20 mL) and H2O (30 mL). The organic phase was dried in vacuo and the crude product used in the subsequent reaction without further purification.
3.2.2.1. N-n-Butyl-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6a)
From N-n-butyl-8-hydroxyquinoline-2-carboxamide (5a) (48 mg, 0.20 mmol), PIFA (254 mg, 0.59 mmol) to give 6a (44 mg, 85% yield) as a brown oil.
1H NMR (CDCl3, 400 MHz) δH 8.59 (1H, d, J = 8.0 Hz, H-3*), 8.56 (1H, d, J = 8.0 Hz, H-4*), 8.28 (1H, br s, NH-2′), 7.19 (1H, d, J = 10.4 Hz, H-7), 7.11 (1H, d, J = 10.4 Hz, H-6), 3.50 (2H, dt, J = 6.4, 6.4 Hz, H2-3′), 1.64 (2H, p, J = 7.3 Hz, H2-4′), 1.41 (2H, sex., J = 7.2 Hz, H2-5′), 0.94 (3H, t, J = 7.6 Hz, H3-6′); 13C NMR (CDCl3, 100 MHz) δC 183.8 (C-5), 182.6 (C-8), 162.4 (C-1′), 153.9 (C-2), 145.8 (C-8a), 139.3 (C-7*), 138.3 (C-6*), 136.5 (C-3*), 130.2 (C-4a), 126.2 (C-4*), 39.6 (C-3′), 31.6 (C-4′), 20.1 (C-5′), 13.8 (C-6′); (+)-ESIMS m/z 281 [M + Na]+; (+)-HRESIMS m/z 281.0893 [M + Na]+ (calcd. for C14H14N2NaO3, 281.0897).
3.2.2.2. N-n-Pentyl-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6b)
From N-n-pentyl-8-hydroxyquinoline-2-carboxamide (5b) (38 mg, 0.15 mmol), PIFA (127 mg, 0.30 mmol) to give 6b (31 mg, 76% yield) as a brown oil.
1H NMR (CDCl3, 300 MHz) δH 8.57 (2H, s, H-3/H-4), 8.30 (1H, br s, NH-2′), 7.20 (1H, d, J = 10.3 Hz, H-7), 7.12 (1H, d, J = 10.3 Hz, H-6), 3.53–3.44 (2H, m, H2-3′), 1.71–1.61 (2H, m, H2-4′), 1.39–1.33 (4H, m, H2-5′/H2-6′), 0.90 (3H, t, J = 7.3 Hz, H3-7′); 13C NMR (CDCl3, 75 MHz) δC 183.7 (C-5*), 182.4 (C-8*), 162.4 (C-1′), 154.0 (C-2), 145.7 (C-8a), 139.2 (C-7), 138.2 (C-6), 136.3 (C-3*), 130.2 (C-4a), 126.1 (C-4*), 39.7 (C-3′), 29.2 (C-4′), 29.0 (C-5′), 22.2 (C-6′), 13.9 (C-7′); (+)-ESIMS m/z 295 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 295.1061 (calcd. for C15H16NaN2O3, 295.1053).
3.2.2.3. N-n-Octyl-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6c)
From N-n-octyl-8-hydroxyquinoline-2-carboxamide (5c) (73 mg, 0.24 mmol), PIFA (127 mg, 0.30 mmol) to give 6c (64 mg, 85% yield) as a brown oil.
1H NMR (CDCl3, 300 MHz) δH 8.56 (2H, s, H-3/H-4), 8.23 (1H, br s, NH-2′), 7.18 (1H, d, J = 10.5 Hz, H-7), 7.10 (1H, d, J = 10.5 Hz, H-6), 3.48 (2H, dt, J = 6.7, 6.0 Hz, H2-3′), 1.65 (2H, p, J = 7.2 Hz, H2-4′), 1.42–1.20 (10H, br s, H2-5′/H2-6′/H2-7′/H2-8′/H2-9′), 0.86 (3H, t, J = 7.1 Hz, H3-10′); 13C NMR (CDCl3, 75 MHz) δC 183.8 (C-5), 182.5 (C-8), 162.4 (C-1′), 154.1 (C-2), 145.0 (C-8a), 139.3 (C-7), 138.2 (C-6), 136.4 (C-3*), 130.3 (C-4a), 126.2 (C-4*), 39.9 (C-3′), 31.8 (C-6′*), 29.6 (C-4′), 29.2 (C-7′*), 29.1 (C-8′*), 27.0 (C-5′), 22.6 (C-9′*), 14.0 (C-10′); (+)-ESIMS m/z 337 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 337.1531 (calcd. for C18H22N2NaO3, 337.1523).
3.2.2.4. N-Benzyl-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6d)
From N-benzyl-8-hydroxyquinoline-2-carboxamide (5d) (51 mg, 0.18 mmol), PIFA (127 mg, 0.30 mmol) to give 6d (44 mg, 84% yield) as a brown oil.
1H NMR (CDCl3, 400 MHz) δH 8.67 (1H, br s, NH-2′), 8.60 (1H, d, J = 8.1 Hz, H-3*), 8.55 (1H, d, J = 8.1 Hz, H-4*), 7.36–7.26 (5H, m, 2H-5′/2H-6′/H-7′), 7.14 (1H, d, J = 10.6 Hz, H-7), 7.09 (1H, d, J = 10.6 Hz, H-6), 4.69 (2H, d, J = 6.3 Hz, H2-3′); 13C NMR (CDCl3, 100 MHz) δC 183.7 (C-5), 182.4 (C-8), 162.5 (C-1′), 153.8 (C-2), 145.8 (C-8a), 139.2 (C-7), 138.2 (C-6), 137.4 (C-4′), 136.4 (C-3*), 130.2 (C-4a), 128.7 (C-5′), 127.8 (C-6′), 127.5 (C-7′), 126.4 (C-4*), 43.6 (C-3′); (+)-ESIMS m/z 315 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 315.0748 (calcd. for C17H12N2NaO3, 315.0740).
3.2.2.5. N-Phenethyl-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6e)
From N-phenethyl-8-hydroxyquinoline-2-carboxamide (5e) (58 mg, 0.20 mmol), PIFA (127 mg, 0.30 mmol) to give 6e (42 mg, 69% yield) as a brown oil.
1H NMR (CDCl3, 300 MHz) δH 8.57 (2H, s, H-3/H-4), 8.32 (1H, br s, NH-2′), 7.35–7.22 (5H, m, 2H-6′/2H-7′/H-8′), 7.17 (1H, d, J = 10.5 Hz, H-7), 7.10 (1H, d, J = 10.5 Hz, H-6), 3.75 (2H, dt, J = 7.7, 6.9 Hz, H2-3′), 2.98 (2H, t, J = 7.7 Hz, H2-4′); 13C NMR (CDCl3, 100 MHz) δC 183.7 (C-5), 182.3 (C-8), 162.4 (C-1′), 153.9 (C-2), 145.8 (C-8a), 139.3 (C-7), 138.2 (C-6), 137.4 (C-5′), 136.4 (C-3*), 130.2 (C-4a), 128.7 (C-6′*), 128.6 (C-7′*), 126.5 (C-8′), 126.1 (C-4*), 41.2 (C-3′), 35.8 (C-4′); (+)-ESIMS m/z 329 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 329.0904 (calcd. for C18H14N2NaO3, 329.0897).
3.2.2.6. N-(3-Phenpropyl)-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6f)
From N-(3-phenylpropyl)-8-hydroxyquinoline-2-carboxamide (5f) (65 mg, 0.21 mmol), PIFA (127 mg, 0.30 mmol) to give 6f (45 mg, 67% yield) as a brown oil.
1H NMR (CDCl3, 400 MHz) δH 8.57 (2H, s, H-3/H-4), 8.34 (1H, br s, NH-2′), 7.33–7.19 (5H, m, 2H-7′/2H-8′/H-9′), 7.17 (1H, d, J = 10.4 Hz, H-7), 7.10 (1H, d, J = 10.4 Hz, H-6), 3.54 (2H, dt, J = 7.5, 6.8 Hz, H2-3′), 2.72 (2H, t, J = 7.5 Hz, H2-5′), 2.01 (2H, p, J = 7.5 Hz, H2-4′); 13C NMR (CDCl3, 100 MHz) δC 183.7 (C-5), 182.4 (C-8), 162.4 (C-1′), 153.9 (C-2), 145.7 (C-8a), 139.2 (C-7), 138.2 (C-6), 137.4 (C-6′), 136.4 (C-3*), 130.2 (C-4a), 128.4 (C-7′), 128.3 (C-8′), 126.1 (C-9′), 125.9 (C-4*), 39.3 (C-3′), 33.2 (C-5′), 31.0 (C-4′); (+)-ESIMS m/z 343 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 343.1063 (calcd. for C19H16N2NaO3, 343.1053).
3.2.2.7. N-Geranyl-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6g)
From N-geranyl-8-hydroxyquinoline-2-carboxamide (5g) (48 mg, 0.15 mmol), PIFA (127 mg, 0.30 mmol) to give 6g (35 mg, 69% yield) as a brown oil.
1H NMR (CDCl3, 400 MHz) δH 8.57 (1H, d, J = 8.1 Hz, H-3), 8.54 (1H, d, J = 8.1 Hz, H-4), 8.18 (1H, br s, NH-2′), 7.19 (1H, d, J = 10.5 Hz, H-7), 7.09 (1H, d, J = 10.5 Hz, H-6), 5.28 (1H, t, J = 5.6 Hz, H-4′), 5.06 (1H, t, J = 6.8 Hz, H-8′), 4.10 (2H, dd, J = 6.4, 5.6 Hz, H2-3′), 2.11–2.05 (2H, m, H2-7′), 2.04–1.99 (2H, m, H2-6′), 1.72 (3H, s, H3-11′), 1.65 (3H, s, H3-12′), 1.58 (3H, s, H3-10′); 13C NMR (CDCl3, 100 MHz) δC 183.8 (C-5), 182.5 (C-8), 162.2 (C-1′), 154.1 (C-2), 145.8 (C-8a), 140.0 (C-5′), 139.3 (C-7), 138.2 (C-6), 136.3 (C-3*), 131.7 (C-9′), 130.3 (C-4a), 126.2 (C-4*), 123.8 (C-8′), 119.4 (C-4′), 39.5 (C-6′), 37.7 (C-3′), 26.3 (C-7′), 25.6 (C-12′), 17.7 (C-10′), 16.4 (C-11′); (+)-ESIMS m/z 361 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 361.1526 (calcd. for C20H22N2NaO3, 361.1523).
3.2.2.8. N-Propargyl-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6h)
From N-propargyl-8-hydroxyquinoline-2-carboxamide (5h) (45 mg, 0.20 mmol), PIFA (229 mg, 0.53 mmol) to give 6h (40 mg, 83% yield) as a brown oil.
1H NMR (CDCl3, 400 MHz) δH 8.57 (2H, br s, H-3/H-4), 7.19 (1H, d, J = 10.5 Hz, H-7), 7.12 (1H, d, J = 10.5 Hz, H-6), 4.30 (2H, dd, J = 5.6, 2.5 Hz, H2-3′), 2.28 (1H, t, J = 2.5 Hz, H-5′); 13C NMR (CDCl3, 100 MHz) δC 183.6 (C-5), 182.4 (C-8), 162.3 (C-1′), 153.3 (C-2), 145.8 (C-8a), 139.3 (C-6*), 138.3 (C-7*), 136.5 (C-3*), 130.2 (C-4a), 126.4 (C-4*), 78.9 (C-4′), 71.8 (C-5′), 29.3 (C-3′); (+)-ESIMS m/z 263 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 263.0431 (calcd. for C13H8N2NaO3, 263.0427).
3.2.2.9. N-(2-Methoxyethyl)-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6i)
From N-(2-methoxyethyl)-8-hydroxyquinoline-2-carboxamide (5i) (86 mg, 0.35 mmol), PIFA (300 mg, 0.70 mmol) to give 6i (78 mg, 86% yield) as a brown oil.
1H NMR (CDCl3, 400 MHz) δH 8.58 (2H, s, H-3/H-4), 8.49 (1H, br s, NH-2′), 7.19 (1H, d, J = 10.4 Hz, H-7), 7.10 (1H, d, J = 10.4 Hz, H-6), 3.72 (2H, dt, J = 5.6, 5.2 Hz, H2-3′), 3.61 (2H, t, J = 5.2 Hz, H2-4′), 3.40 (3H, s, H3-5′); 13C NMR (CDCl3, 100 MHz) δC 183.8 (C-5), 182.3 (C-8), 162.7 (C-1′), 153.9 (C-2), 145.9 (C-8a), 139.3 (C-7*), 138.2 (C-6*), 136.4 (C-3*), 130.2 (C-4a), 126.2 (C-4*), 70.9 (C-4′), 58.9 (C-5′), 39.6 (C-3′); (+)-ESIMS m/z 283 [M + Na]+; (+)-HRESIMS m/z 283.0696 [M + Na]+ (calcd. for C13H12N2NaO4, 283.0689).
3.2.2.10. N-Glycine(methylester)-5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6j)
From N-glycine(methylester)-8-hydroxyquinoline-2-carboxamide (5j) (50 mg, 0.19 mmol), PIFA (132 mg, 0.31 mmol) to give 6j (40 mg, 77% yield) as a brown oil.
1H NMR (CDCl3, 400 MHz) δH 8.77 (2H, br t, J = 5.2 Hz, NH-2′), 8.59 (1H, d, J = 8.3 Hz, H-3), 8.55 (1H, d, J = 8.3 Hz, H-4), 7.20 (1H, d, J = 10.5 Hz, H-7), 7.13 (1H, d, J = 10.5 Hz, H-6), 4.31 (2H, d, J = 6.1 Hz, H2-3′), 3.79 (3H, s, H3-5′); 13C NMR (CDCl3, 100 MHz) δC 183.7 (C-5), 182.4 (C-8), 169.7 (C-4′), 163.1 (C-1′), 153.2 (C-2), 145.9 (C-8a), 139.4 (C-7), 138.3 (C-6), 136.5 (C-4), 130.2 (C-4a), 126.4 (C-3), 52.5 (C-5′), 41.4 (C-3′); (+)-ESIMS m/z 297 [M + Na]+; (+)-HRESIMS m/z 297.0477 [M + Na]+ (calcd. for C13H10N2NaO5, 297.0482).
3.2.3. General Procedure for the Preparation of Carboxamide Analogues 7a–7j
A solution of 5,8-dioxo-5,8-dihydroquinoline-2-carboxamide (6a–6j) was dissolved in MeCN/EtOH (1:1) before being cooled to 0 °C. In some cases, CeCl3·7H2O (1 equiv.) was also added to the reaction. Hypotaurine (0.8 equiv.) in H2O was added dropwise over 3.5 h. The reaction mixture changed color from dark brown to dark orange, and was stirred at rt for 2 days. The product was purified either by filtration and washing with H2O (3 × 20 mL) and MeOH (3 × 20 mL), or by reversed-phase C18 flash column chromatography (0%–80% MeOH in H2O (0.05% TFA)).
3.2.3.1. N-n-Butyl-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (7a)
From 6a (54 mg, 0.21 mmol) in MeCN/EtOH (1:1, 20 mL) and hypotaurine (16.0 mg, 0.15 mmol) in H2O (3 mL). Filtration gave 7a as an orange powder (11.0 mg, 14% yield).
Mp 200 °C (decomp.); Rf = 0.49 (10% MeOH/CH2Cl2); IR νmax (ATR) 3300, 3237, 1682, 1594, 1580, 1508, 1336, 1280, 1170, 1107 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 9.35 (1H, br s, NH-4), 8.70 (1H, t, J = 6.4 Hz, NH-2′), 8.53 (1H, d, J = 8.2 Hz, H-9), 8.40 (1H, d, J = 8.2 Hz, H-8), 3.92–3.87 (2H, m, H2-3), 3.43–3.36 (obscured by solvent, H2-2 and H2-3′), 1.55 (2H, p, J = 7.2 Hz, H2-4′), 1.33 (2H, sex., J = 7.6 Hz, H2-5′), 0.91 (3H, t, J = 7.6 Hz, H3-6′); 13C NMR (DMSO-d6, 100 MHz) δC 176.2 (C-5), 173.4 (C-10), 162.5 (C-1′), 152.6 (C-7), 147.7 (C-4a), 145.3 (C-5a), 136.0 (C-9), 131.4 (C-9a), 126.5 (C-8), 110.7 (C-10a), 48.2 (C-2), 39.3 (obscured by solvent, C-3 and C-3′), 31.3 (C-4′), 19.6 (C-5′), 13.7 (C-6′); (+)-ESIMS m/z 386 [M + Na]+; (+)-HRESIMS m/z 386.0791 [M + Na]+ (calcd. for C16H17N3NaO5S, 386.0781).
3.2.3.2. N-n-Pentyl-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (7b)
From 6b (31 mg, 0.11 mmol), CeCl3.7H2O (37 mg, 98 µmol) in MeCN/EtOH (1:1, 14 mL) and hypotaurine (8.2 mg, 0.075 mmol) in H2O (2 mL). Filtration gave 7b as a red-brown powder (11.0 mg, 27% yield).
Mp 200 °C (decomp.); Rf = 0.44 (10% MeOH/CH2Cl2); IR νmax (ATR) 3234, 2933, 1686, 1508 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 9.36 (1H, br s, NH-4), 8.72 (1H, t, J = 5.9 Hz, NH-2′), 8.53 (1H, d, J = 8.2 Hz, H-9), 8.40 (1H, d, J = 8.2 Hz, H-8), 3.90 (2H, br s, H2-3), 3.43–3.33 (4H, obscured by H2O, H2-2, H2-3′), 1.57 (2H, p, J = 6.8 Hz, H2-4′), 1.34–1.27 (4H, m, H2-5′/H2-6′), 0.88 (3H, t, J = 6.8 Hz, H3-7′); 13C NMR (DMSO-d6, 100 MHz) δC 176.2 (C-5), 173.5 (C-10), 162.5 (C-1′), 152.6 (C-7), 147.7 (C-4a), 145.4 (C-5a), 136.0 (C-9), 131.4 (C-9a), 126.5 (C-8), 110.7 (C-10a), 48.2 (C-2), 40.8 (C-3), 38.8 (C-3′), 28.8 (C-4′), 28.7 (C-5′*), 21.9 (C-6′*), 13.9 (C-7′); (+)-ESIMS m/z 378 [M + H]+; (+)-HRESIMS m/z [M + H]+ 378.1107 (calcd. for C17H20N3O5S, 378.1118).
3.2.3.3. N-n-Octyl-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (7c)
From 6c (32 mg, 0.10 mmol), CeCl3·7H2O (39 mg, 0.10 mmol) in MeCN/EtOH (1:1, 14 mL) and hypotaurine (9.7 mg, 0.089 mmol) in H2O (2 mL). Filtration and solvent wash gave 7c as a red-brown powder (24.0 mg, 57% yield).
Mp 200 °C (decomp.); Rf = 0.44 (10% MeOH/CH2Cl2); IR νmax (ATR) 3240, 2925, 1669, 1521 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 9.36 (1H, br s, NH-4), 8.71 (1H, t, J = 6.1 Hz, NH-2′), 8.53 (1H, d, J = 8.0 Hz, H-9), 8.39 (1H, d, J = 8.0 Hz, H-8), 3.90 (2H, br s, H2-3), 3.41 (2H, br t, J = 6.1 Hz, H2-2), 3.32 (2H, obscured by H2O, H2-3′), 1.61–1.53 (2H, m, H2-4′), 1.37–1.20 (10H, m, H2-5′/H2-6′/H2-7′/H2-8′/H2-9′), 0.85 (3H, t, J = 6.7 Hz, H3-10′); 13C NMR (DMSO-d6, 100 MHz) δC 176.1 (C-5), 173.4 (C-10), 162.4 (C-1′), 152.5 (C-7), 147.6 (C-4a), 145.2 (C-5a), 135.9 (C-9), 131.3 (C-9a), 126.4 (C-8), 110.6 (C-10a), 48.1 (C-2), 39.5 (C-3/C-3′), 31.1 (C-8′), 29.0 (C-4′), 28.6 (C-6′), 26.4 (C-5′), 22.0 (C-9′), 13.9 (C-10′); (+)-ESIMS m/z 420 [M + H]+; (+)-HRESIMS m/z [M + H]+ 420.1581 (calcd. for C20H26N3O5S, 420.1588).
3.2.3.4. N-Benzyl-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (7d)
From 6d (44 mg, 0.15 mmol), CeCl3.7H2O (55 mg, 0.15 mmol) in MeCN/EtOH (1:1, 14 mL) and hypotaurine (13.0 mg, 0.12 mmol) in H2O (2 mL). Filtration and solvent wash gave 7d as a red-brown powder (10.0 mg, 17% yield).
Mp 200 °C (decomp.); Rf = 0.44 (10% MeOH/CH2Cl2); IR νmax (ATR) 3267, 1676, 1595, 1513 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 9.37 (1H, br s, NH-4), 9.28 (1H, t, J = 6.2 Hz, NH-2′), 8.55 (1H, d, J = 8.2 Hz, H-9), 8.43 (1H, d, J = 8.2 Hz, H-8), 7.37–7.23 (5H, m, 2H-5′/2H-6′/H-7′), 4.57 (2H, d, J = 6.4 Hz, H2-3′), 3.90 (2H, br s, H2-3), 3.41 (2H, t, J = 6.2 Hz, H2-2); 13C NMR (DMSO-d6, 75 MHz) δC 176.2 (C-5), 173.4 (C-10), 162.8 (C-1′), 152.5 (C-7), 147.7 (C-4a), 145.5 (C-5a), 139.3 (C-4′), 136.0 (C-9), 131.5 (C-9a), 128.4 (C-5′), 127.5 (C-6′), 126.9 (C-8), 126.7 (C-7′), 110.7 (C-10a), 48.2 (C-2), 42.7 (C-3′), 39.4 (C-3); (+)-ESIMS m/z 420 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 420.0618 (calcd. for C19H15N3NaO5S, 420.0625).
3.2.3.5. N-Phenethyl-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (7e)
From 6e (22.8 mg, 0.075 mmol), CeCl3.7H2O (51 mg, 0.14 mmol) in MeCN/EtOH (1:1, 14 mL) and hypotaurine (12.0 mg, 0.11 mmol) in H2O (2 mL). Filtration and solvent wash gave 7e as a red-brown powder (15.0 mg, 49% yield).
Mp 240 °C (decomp.); Rf = 0.48 (10% MeOH/CH2Cl2); IR νmax (ATR) 3230, 1678, 1580, 1555 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 9.37 (1H, br s, NH-4), 8.77 (1H, t, J = 5.9 Hz, NH-2′), 8.54 (1H, d, J = 8.1 Hz, H-9), 8.40 (1H, d, J = 8.1 Hz, H-8), 7.33–7.19 (5H, m, 2H-6′/2H-7′/H-8′), 3.93–3.87 (2H, m, H2-3), 3.60 (2H, dt, J = 7.7, 5.9 Hz, H2-3′), 3.40 (2H, br t, J = 5.4 Hz, H2-2), 2.90 (2H, t, J = 7.7 Hz, H2-4′); 13C NMR (DMSO-d6, 75 MHz) δC 176.2 (C-5), 173.4 (C-10), 162.5 (C-1′), 152.4 (C-7), 147.7 (C-4a), 145.3 (C-5a), 139.2 (C-5′), 136.0 (C-9), 131.4 (C-9a), 128.6 (C-6′), 128.4 (C-7′), 126.5 (C-8′), 126.2 (C-8), 110.7 (C-10a), 48.2 (C-2), 40.7 (C-3′), 38.6 (C-3), 35.1 (C-4′); (+)-ESIMS m/z 434 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 434.0768 (calcd. for C20H17N3NaO5S, 434.0781).
3.2.3.6. N-(3-Phenylpropyl)-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino[2,3-g] quinoline-7-carboxamide 1,1-Dioxide (7f)
From 6f (39.0 mg, 0.12 mmol), CeCl3.7H2O (51.0 mg, 0.14 mmol) in MeCN and EtOH (1:1, 14 mL) and hypotaurine (12.0 mg, 0.11 mmol) in H2O (2 mL). Filtration and solvent wash gave 7f as a red-brown powder (29.0 mg, 57% yield).
Mp 204 °C (decomp.); Rf = 0.52 (10% MeOH/CH2Cl2); IR νmax (ATR) 3247, 2922, 1670, 1528 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 9.36 (1H, br s, NH-4), 8.77 (1H, t, J = 6.0 Hz, NH-2′), 8.54 (1H, d, J = 8.1 Hz, H-9), 8.40 (1H, d, J = 8.1 Hz, H-8), 7.31–7.15 (5H, m, H-7′/H-8′/H-9′), 3.90 (2H, t, J = 5.5 Hz, H2-3), 3.44–3.35 (4H, m, H2-2/H2-3′), 2.64 (2H, t, J = 7.8 Hz, H2-5′), 1.89 (2H, p, J = 7.8 Hz, H2-4′); 13C NMR (DMSO-d6, 75 MHz) δC 176.2 (C-5), 173.5 (C-10), 162.6 (C-1′), 152.6 (C-7), 147.7 (C-4a), 145.4 (C-5a), 141.6 (C-6′), 136.0 (C-9), 131.4 (C-9a), 128.3 (C-7′/C-8′), 126.6 (C-9′), 125.8 (C-8), 110.7 (C-10a), 48.2 (C-2), 39.1 (C-3), 38.8 (C-3′), 32.6 (C-5′), 30.8 (C-4′); (+)-ESIMS m/z 426 [M + H]+; (+)-HRESIMS m/z [M + H]+ 426.1119 (calcd. for C21H20N3O5S, 426.1118).
3.2.3.7. (E)-N-(3,7-Dimethylocta-2,6-dien-1-yl)-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino [2,3-g]quinoline-7-carboxamide 1,1-Dioxide (7g)
From 6g (26.6 mg, 0.079 mmol), CeCl3.7H2O (31 mg, 0.083 mmol) in MeCN/EtOH (1:1, 14 mL) and hypotaurine (7.2 mg, 0.066 mmol) in H2O (2 mL). Filtration and solvent wash gave 7g as a dark orange powder (10.0 mg, 29% yield).
Mp 200 °C (decomp.); Rf = 0.45 (10% MeOH/CH2Cl2); IR νmax (ATR) 3230, 3076, 1693, 1561 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 9.35 (1H, br s, NH-4), 8.73 (1H, t, J = 5.7 Hz, NH-2′), 8.53 (1H, d, J = 8.1 Hz, H-9), 8.41 (1H, d, J = 8.1 Hz, H-8), 5.27 (1H, t, J = 6.2 Hz, H-4′), 5.07 (1H, t, J = 6.2 Hz, H-8′), 3.97 (2H, dd, J = 6.2, 5.7 Hz, H2-3′), 3.90 (2H, br t, J = 5.5 Hz, H2-3), 3.41 (2H, t, J = 5.5 Hz, H2-2), 2.09–2.02 (2H, m, H2-7′), 2.01–1.95 (2H, m, H2-6′), 1.71 (3H, s, H3-11′), 1.62 (3H, s, H3-12′), 1.56 (3H, s, H3-10′); 13C NMR (DMSO-d6, 75 MHz) δC 176.2 (C-5), 173.4 (C-10), 162.3 (C-1′), 152.6 (C-7), 147.7 (C-4a), 145.3 (C-5a), 137.5 (C-5′), 136.0 (C-9), 131.4 (C-9a), 130.9 (C-9′), 126.5 (C-8), 123.9 (C-8′), 121.0 (C-4′), 110.7 (C-10a), 48.2 (C-2), 40.3 (C-3), 38.6 (C-6′), 37.1 (C-3′), 25.9 (C-7′), 25.5 (C-12′), 17.5 (C-10′), 16.1 (C-11′); (+)-ESIMS m/z 466 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 466.1395 (calcd. for C22H25N3NaO5S, 466.1407).
3.2.3.8. N-(Prop-2-yn-1-yl)-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (7h)
From 6h (35 mg, 0.15 mmol), CeCl3.7H2O (46.5 mg, 0.12 mmol) in MeCN/EtOH (1:1, 14 mL) and hypotaurine (9.5 mg, 0.087 mmol) in H2O (2 mL). The crude reaction mixture was purified by reversed-phase C18 flash column chromatography to give 7h as a bright yellow powder (15.0 mg, 29% yield).
Mp 280 °C (decomp.); Rf = 0.52 (10% MeOH/CH2Cl2); IR νmax (ATR) 3369, 3255, 2936, 1667, 1595 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 9.38 (1H, t, J = 5.4 Hz, NH-4), 9.09 (1H, t, J = 6.0 Hz, NH-2′), 8.55 (1H, d, J = 8.1 Hz, H-9), 8.41 (1H, d, J = 8.1 Hz, H-8), 4.13 (2H, dd, J = 6.0, 2.4 Hz, H2-3′), 3.93–3.87 (2H, m, H2-3), 3.40 (2H, obscured by water, H2-2), 3.12 (1H, t, J = 2.4 Hz, H-5′); 13C NMR (DMSO-d6, 75 MHz) δC 176.1 (C-5), 173.3 (C-10), 162.6 (C-1′), 152.0 (C-7), 147.7 (C-4a), 145.5 (C-5a), 136.0 (C-9), 131.5 (C-9a), 126.7 (C-8), 110.7 (C-10a), 80.9 (C-4′), 72.8 (C-5′), 48.2 (C-2), 39.4 (C-3), 28.7 (C-3′); (+)-ESIMS m/z 368 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 368.0294 (calcd. for C15H11N3NaO5S, 368.0312).
3.2.3.9. N-(2-Methoxyethyl)-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino[2,3-g] quinoline-7-carboxamide 1,1-Dioxide (7i)
From 6i (31 mg, 0.12 mmol) in MeCN/EtOH (1:1, 20 mL) and hypotaurine (7.8 mg, 0.072 mmol) in H2O (3 mL). The crude reaction mixture was purified by reversed-phase C18 flash column chromatography to give 7i as an orange powder (11.2 mg, 26% yield).
Mp 200 °C (decomp.); Rf = 0.54 (10% MeOH/CH2Cl2); IR νmax (ATR) 3546, 3251, 1673, 1594, 1581, 1556, 1339, 1122 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 9.37 (1H, br s, NH-4), 8.64 (1H, t, J = 5.4 Hz, NH-2′), 8.54 (1H, d, J = 8.0 Hz, H-9), 8.41 (1H, d, J = 8.0 Hz, H-8), 3.92–3.88 (2H, m, H2-3), 3.56–3.51 (4H, m, H2-3′ and H2-4′), 3.43–3.39 (2H, m, H2-2), 3.29 (3H, s, H3-5′); 13C NMR (DMSO-d6, 100 MHz) δC 176.2 (C-5), 173.4 (C-10), 162.5 (C-1′), 152.2 (C-7), 147.7 (C-4a), 145.4 (C-5a), 136.1 (C-9), 131.4 (C-9a), 126.5 (C-8), 110.7 (C-10a), 70.3 (C-4′), 57.9 (C-5′), 48.2 (C-2), 39.2 (C-3), 38.7 (C-3′); (+)-ESIMS m/z 388 [M + Na]+; (+)-HRESIMS m/z 388.0566 [M + Na]+ (calcd. for C15H15N3NaO6S, 388.0574).
3.2.3.10. Methyl 2-(1,1-Dioxido-5,10-dioxo-3,4,5,10-tetrahydro-2H-[1,4]thiazino[2,3-g] quinoline-7-carboxamido)acetate (7j)
From 6j (50 mg, 0.18 mmol) in MeCN/EtOH (1:1, 20 mL) and hypotaurine (11.9 mg, 0.11 mmol) in H2O (3 mL). Reversed-phase C18 flash column chromatography gave 7j as a bright red powder (13.8 mg, 20% yield).
Mp 200 °C (decomp.); Rf = 0.46 (10% MeOH/CH2Cl2); IR νmax (ATR) 3576, 3335, 1748, 1666, 1594, 1581, 1557, 1346, 1271, 1212, 1164, 1115 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 9.40 (1H, br t, J = 3.4 Hz, NH-4), 9.08 (1H, t, J = 6.1 Hz, NH-2′), 8.56 (1H, d, J = 8.0 Hz, H-9), 8.42 (1H, d, J = 8.0 Hz, H-8), 4.15 (2H, d, J = 6.1 Hz, H-3′), 3.92–3.88 (2H, m, H2-3), 3.67 (3H, s, H3-5′), 3.43–3.40 (2H, m, H2-2); 13C (DMSO-d6, 100 MHz) δC 176.2 (C-5), 173.4 (C-10), 170.0 (C-4′), 163.0 (C-1′), 151.7 (C-7), 147.7 (C-4a), 145.6 (C-5a), 136.1 (C-9), 131.6 (C-9a), 126.6 (C-8), 110.8 (C-10a), 51.9 (C-5′), 48.2 (C-2), 41.2 (C-3′), 39.2 (C-3); (+)-ESIMS m/z 380 [M + H]+; (+)-HRESIMS m/z 380.0538 [M + H]+ (calcd. for C15H14N3O7S, 380.0547).
3.2.4. General Procedure for Preparation of Δ2(3) Analogues 8a–8i, 8k
Thiazine-quinoline-carboxamide (7a–7j) in DMF (1–3 mL) was stirred in 2 N NaOH (3 mL) at rt for 2 h. HCl (10% vol) was added dropwise until the reaction mixture was pH 5 and the mixture was then purified by reversed-phase C18 flash column chromatography (0%–10% MeOH (0.05% TFA)) to give the desired product.
3.2.4.1. N-n-Butyl-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (8a)
From 7a (34.0 mg, 0.094 mmol) using the general procedure to give 8a as a yellow solid (13.0 mg, 38% yield).
Mp 200 °C (decomp.); Rf = 0.53 (10% MeOH/CH2Cl2); IR νmax (ATR) 3402, 3058, 1714, 1653, 1632, 1527, 1503, 1318, 1125 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 11.42 (1H, br s, NH-4), 8.76 (1H, t, J = 6.0 Hz, NH-2′), 8.58 (1H, d, J = 8.0 Hz, H-9), 8.43 (1H, d, J = 8.0 Hz, H-8), 7.17 (1H, d, J = 9.0 Hz, H-3), 6.62 (1H, d, J = 9.0 Hz, H-2), 3.38 (2H, dt, J = 6.9, 6.9 Hz, H2-3′), 1.56 (2H, p, J = 7.0 Hz, H2-4′), 1.33 (2H, sex., J = 7.5 Hz, H2-5′), 0.91 (3H, t, J = 7.5 Hz, H3-6′); 13C NMR (DMSO-d6, 100 MHz) δC 177.7 (C-10), 175.5 (C-5), 162.4 (C-1′), 153.3 (C-7), 145.5 (C-5a), 141.4 (C-4a), 136.1 (C-9), 130.6 (C-9a), 130.5 (C-3), 126.4 (C-8), 115.2 (C-10a), 112.0 (C-2), 38.6 (C-3′), 31.3 (C-4′), 19.6 (C-5′), 13.7 (C-6′); (+)-ESIMS m/z 384 [M + Na]+; (+)-HRESIMS m/z 384.0632 [M + Na]+ (calcd. for C16H15N3NaO5S, 384.0625).
3.2.4.2. N-n-Pentyl-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (8b)
From 7b (10.0 mg, 0.027 mmol) using the general procedure to give 8b as a yellow solid (4.0 mg, 40% yield).
Mp 280 °C (decomp.); Rf = 0.44 (10% MeOH/CH2Cl2); IR νmax (ATR) 3319, 3057, 1710, 1635, 1528 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 11.44 (1H, br s, NH-4), 8.78 (1H, t, J = 6.1 Hz, NH-2′), 8.58 (1H, d, J = 8.1 Hz, H-9), 8.43 (1H, d, J = 8.1 Hz, H-8), 7.17 (1H, d, J = 8.9 Hz, H-3), 6.62 (1H, d, J = 8.9 Hz, H-2), 3.40–3.34 (2H, m, H2-3′), 1.58 (2H, p, J = 7.5 Hz, H2-4′), 1.34–1.27 (4H, m, H2-5′/H2-6′), 0.88 (3H, t, J = 7.5 Hz, H3-7′); 13C NMR (DMSO-d6, 75 MHz) δC 177.7 (C-10), 175.5 (C-5), 162.4 (C-1′), 153.3 (C-7), 145.6 (C-5a), 141.4 (C-4a), 136.1 (C-9), 130.6 (C-9a), 130.5 (C-3), 126.4 (C-8), 115.2 (C-10a), 112.0 (C-2), 38.8 (C-3′), 28.9 (C-4′), 28.7 (C-5′), 21.9 (C-6′), 14.0 (C-7′); (+)-ESIMS m/z 398 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 398.0776 (calcd. for C17H17N3NaO5S, 3798.0781).
3.2.4.3. N-n-Octyl-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (8c)
From 7c (12.0 mg, 0.029 mmol) using the general procedure to give 8c as a yellow solid (6.0 mg, 50% yield).
Mp 280 °C (decomp.); Rf = 0.41 (10% MeOH/CH2Cl2); IR νmax (ATR) 3289, 2924, 1635, 1527 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 11.44 (1H, d, J = 5.3 Hz, NH-4), 8.79 (1H, t, J = 5.8 Hz, NH-2′), 8.58 (1H, d, J = 8.0 Hz, H-9), 8.43 (1H, d, J = 8.0 Hz, H-8), 7.17 (1H, dd, J = 8.8, 5.3 Hz, H-3), 6.63 (1H, d, J = 8.8 Hz, H-2), 3.37 (obscured by solvent, H2-3′), 1.60–1.52 (2H, m, H2-4′), 1.31–1.23 (10H, m, H2-5′/H2-6′/H2-7′/H2-8′/H2-9′), 0.85 (3H, t, J = 6.8 Hz, H3-10′); 13C NMR (DMSO-d6, 75 MHz) δC 177.7 (C-10), 175.5 (C-5), 162.4 (C-1′), 153.3 (C-7), 145.6 (C-5a), 141.4 (C-4a), 136.1 (C-9), 130.6 (C-9a), 130.5 (C-3), 126.4 (C-8), 115.2 (C-10a), 112.0 (C-2), 38.9 (C-3′), 31.3 (C-8′), 29.2 (C-4′), 28.8 (C-6′), 28.7 (C-7′), 26.5 (C-5′), 22.1 (C-9′), 14.0 (C-10′); (+)-ESIMS m/z 440 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 440.1232 (calcd. for C20H23N3NaO5S, 440.1251).
3.2.4.4. N-Benzyl-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (8d)
From 7d (10.0 mg, 0.025 mmol) using the general procedure to give 8d as a yellow solid (3.0 mg, 30% yield).
Mp 280 °C (decomp.); Rf = 0.47 (10% MeOH/CH2Cl2); IR νmax (ATR) 3213, 1706, 1634, 1513 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 11.41 (1H, d, J = 5.6 Hz, NH-4), 9.33 (1H, t, J = 6.3 Hz, NH-2′), 8.59 (1H, d, J = 8.2 Hz, H-9), 8.46 (1H, d, J = 8.2 Hz, H-8), 7.38–7.30 (4H, m, 2H-5′/2H-6′), 7.27–7.23 (1H, m, H-7′), 7.17 (1H, dd, J = 8.9, 5.6 Hz, H-3), 6.61 (1H, d, J = 8.9 Hz, H-2), 4.58 (2H, d, J = 6.3 Hz, H2-3′); 13C NMR (DMSO-d6, 100 MHz) δC 177.6 (C-10), 175.4 (C-5), 162.7 (C-1′), 153.1 (C-7), 145.6 (C-5a), 141.3 (C-4a), 139.2 (C-4′), 136.1 (C-9), 130.7 (C-9a), 130.5 (C-3), 128.3 (C-5′), 127.5 (C-6′), 126.9 (C-8), 126.6 (C-7′), 115.2 (C-10a), 112.0 (C-2), 42.7 (C-3′); (+)-ESIMS m/z 418 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 418.0470 (calcd. for C19H13N3NaO5S, 418.0468).
3.2.4.5. N-Phenethyl-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (8e)
From 7e (11.0 mg, 0.027 mmol) using the general procedure to give 8e as a yellow solid (4.0 mg, 36% yield).
Mp 290 °C (decomp.); Rf = 0.47 (10% MeOH/CH2Cl2); IR νmax (ATR) 3103, 3067, 1714, 1678, 1512 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 11.45 (1H, d, J = 5.6, NH-4), 8.85 (1H, t, J = 6.1 Hz, NH-2′), 8.58 (1H, d, J = 8.1 Hz, H-9), 8.44 (1H, d, J = 8.1 Hz, H-8), 7.34–7.22 (5H, m, 2H-6′/2H-7′/H-8′), 7.17 (1H, dd, J = 8.7, 5.6 Hz, H-3), 6.62 (1H, d, J = 8.7 Hz, H-2), 3.61 (2H, dt, J = 6.9, 6.1 Hz, H2-3′), 2.90 (2H, t, J = 6.9 Hz, H2-4′); 13C NMR (DMSO-d6, 100 MHz) δC 177.7 (C-10), 175.5 (C-5), 162.4 (C-1′), 153.0 (C-7), 145.6 (C-5a), 141.4 (C-4a), 139.3 (C-5′), 136.2 (C-9), 130.7 (C-9a), 130.5 (C-3), 128.7 (C-6′), 128.5 (C-7′), 126.4 (C-7′), 126.2 (C-8), 115.2 (C-10a), 112.0 (C-2), 40.8 (C-3′), 35.1 (C-4′); (+)-ESIMS m/z 432 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 432.0618 (calcd. for C20H15N3NaO5S, 432.0625).
3.2.4.6. N-(3-Phenylpropyl)-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (8f)
From 7f (20.0 mg, 0.047 mmol) using the general procedure to give 8f as a yellow solid (6.0 mg, 30% yield).
Mp 230 °C (decomp.); Rf = 0.41 (10% MeOH/CH2Cl2); IR νmax (ATR) 3059, 2930, 1653, 1511 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 11.45 (1H, br s, NH-4), 8.86 (1H, t, J = 6.1 Hz, NH-2′), 8.58 (1H, d, J = 8.2 Hz, H-9), 8.44 (1H, d, J = 8.2 Hz, H-8), 7.31–7.15 (6H, m, H-3/2H-7′/2H-8′/H-9′), 6.63 (1H, d, J = 9.1 Hz, H-2), 3.40 (2H, dt, J = 7.5, 6.1 Hz, H2-3′), 2.64 (2H, t, J = 7.5 Hz, H2-5′), 1.89 (2H, p, J = 7.5 Hz, H2-4′); 13C NMR (DMSO-d6, 100 MHz) δC 177.7 (C-10), 175.5 (C-5), 162.5 (C-1′), 153.3 (C-7), 145.6 (C-5a), 141.6 (C-6′), 141.4 (C-4a), 136.1 (C-9), 130.6 (C-9a), 130.5 (C-3), 128.3 (C-7′/C-8′), 126.5 (C-9′), 125.8 (C-8), 115.2 (C-10a), 112.0 (C-2), 38.9 (C-3′), 32.7 (C-5′), 30.8 (C-4′); (+)-ESIMS m/z 446 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 446.0790 (calcd. for C21H17N3NaO5S, 446.0781).
3.2.4.7. (E)-N-(3,7-Dimethylocta-2,6-dien-1-yl)-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g] quinoline-7-carboxamide 1,1-Dioxide (8g)
From 7g (10.0 mg, 0.023 mmol) using the general procedure to give 8g a yellow solid (5.0 mg, 50% yield).
Mp 280 °C (decomp.); Rf = 0.44 (10% MeOH/CH2Cl2); 1H NMR (DMSO-d6, 400 MHz) δH 11.44 (1H, d, J = 5.7 Hz, NH-4), 8.81 (1H, t, J = 6.2 Hz, NH-2′), 8.58 (1H, d, J = 7.8 Hz, H-9), 8.44 (1H, d, J = 7.9 Hz, H-8), 7.17 (1H, dd, J = 8.6, 5.7 Hz, H-3), 6.62 (1H, d, J = 8.6 Hz, H-2), 5.27 (1H, t, J = 6.6 Hz, H-4′), 5.07 (1H, t, J = 7.0 Hz, H-8′), 3.98 (2H, dd, J = 6.2, 5.8 Hz, H2-3′), 2.07–2.03 (2H, m, H2-6′), 2.01–1.96 (2H, m, H2-7′), 1.71 (3H, s, H3-11′), 1.62 (3H, s, H3-12′), 1.56 (3H, s, H3-10′); 13C NMR (DMSO-d6, 100 MHz) δC 177.6 (C-10), 175.5 (C-5), 162.2 (C-1′), 153.2 (C-7), 145.6 (C-5a), 141.4 (C-4a), 137.6 (C-5′), 136.1 (C-9), 131.0 (C-9a), 130.6 (C-9′), 130.5 (C-3), 126.4 (C-8), 123.9 (C-8′), 121.1 (C-4′), 115.2 (C-10a), 112.0 (C-2), 38.9 (C-6′), 37.1 (C-3′), 26.0 (C-7′), 25.5 (C-12′), 17.6 (C-10′), 16.2 (C-11′); (+)-ESIMS m/z 464 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 464.1254 (calcd. for C22H23N3NaO5S, 464.1251).
3.2.4.8. N-(Prop-2-yn-1-yl)-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g]quinoline-7-carboxamide 1,1-Dioxide (8h)
From 7h (8.0 mg, 0.023 mmol) using the general procedure to give 8h as a yellow solid (6.0 mg, 76% yield).
Mp 230 °C (decomp.); Rf = 0.46 (10% MeOH/CH2Cl2); IR νmax (ATR) 3310, 3058, 1636, 1509 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 11.45 (1H, d, J = 5.5 Hz, NH-4), 9.16 (1H, t, J = 6.2 Hz, NH-2′), 8.60 (1H, d, J = 8.2 Hz, H-9), 8.45 (1H, d, J = 8.2 Hz, H-8), 7.17 (1H, dd, J = 8.8, 5.5 Hz, H-3), 6.62 (1H, d, J = 8.8 Hz, H-2), 4.15 (2H, dd, J = 5.9, 2.5 Hz, H2-3′), 3.13 (1H, t, J = 2.5 Hz, H-5′); 13C NMR (CDCl3, 100 MHz) δC 177.6 (C-10), 175.4 (C-5), 162.5 (C-1′), 152.7 (C-7), 145.8 (C-5a), 141.4 (C-4a), 136.1 (C-9), 130.8 (C-9a), 130.5 (C-3), 126.6 (C-8), 115.3 (C-10a), 112.0 (C-2), 80.9 (C-4′), 72.9 (C-5′), 28.7 (C-3′); (+)-ESIMS m/z 366 [M + Na]+; (+)-HRESIMS m/z [M + Na]+ 366.0151 (calcd. for C15H9N3NaO5S, 366.0155).
3.2.4.9. N-(2-Methoxyethyl)-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g] quinoline-7-carboxamide 1,1-Dioxide (8i)
From 7i (18.8 mg, 0.052 mmol) using the general procedure to give 8i as a yellow solid (15.6 mg, 83% yield).
Mp 200 °C (decomp.); Rf = 0.54 (10% MeOH/CH2Cl2); IR νmax (ATR) 3250, 3057, 1633, 1508, 1278, 1097 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 11.44 (1H, s, NH-4), 8.70 (1H, t, J = 5.6 Hz, H-2′), 8.59 (1H, d, J = 8.0 Hz, H-9), 8.45 (1H, d, J = 8.0 Hz, H-8), 7.17 (1H, d, J = 9.0 Hz, H-3), 6.62 (1H, d, J = 9.0 Hz, H-2), 3.59–3.50 (4H, m, H2-3′ and H2-4′), 3.29 (3H, s, H3-5′); 13C NMR (DMSO-d6, 100 MHz) δC 177.6 (C-10), 175.4 (C-5), 162.4 (C-1′), 152.8 (C-7), 145.5 (C-5a), 141.3 (C-4a), 136.2 (C-9), 130.7 (C-9a), 130.5 (C-3), 126.3 (C-8), 115.3 (C-10a), 112.0 (C-2), 70.2 (C-4′), 57.9 (C-5′), 38.4 (C-3′); (+)-ESIMS m/z 364 [M + H]+; (+)-HRESIMS m/z 364.0606 [M + H]+ (calcd. for C15H14N3O6S, 364.0598).
3.2.4.10. 2-(1,1-Dioxido-5,10-dioxo-5,10-dihydro-4H-[1,4]thiazino[2,3-g]quinoline-7-carboxamido)acetic Acid (8k)
From 7j (13.8 mg, 0.036 mmol) using the general procedure to give carboxylic acid 8k as a yellow oil (8.2 mg, 62% yield).
Rf = 0.20 (10% MeOH/CH2Cl2); IR νmax (ATR) 3582, 3250, 3057, 1748, 1634, 1508, 1279, 1127 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 11.95 (1H, br s, NH-4), 9.00 (1H, t, J = 6.0 Hz, NH-2′), 8.60 (1H, d, J = 8.0 Hz, H-9), 8.45 (1H, d, J = 8.0 Hz, H-8), 7.18 (1H, d, J = 8.8 Hz, H-3), 6.62 (1H, d, J = 8.8 Hz, H-2), 4.07 (2H, d, J = 6.0 Hz, H2-3′); 13C NMR (DMSO-d6, 100 MHz) δC 177.6 (C-10), 175.5 (C-5), 170.9 (C-4′), 162.7 (C-1′), 152.4 (C-7), 145.7 (C-5a), 141.6 (C-4a), 136.2 (C-9), 130.8 (C-9a), 130.6 (C-3), 126.4 (C-8), 115.2 (C-10a), 112.0 (C-2), 41.3 (C-3′); (−)-ESIMS m/z 362 [M − H]−; (−)-HRESIMS m/z 362.0083 [M − H]− (calcd. for C14H8N3O7S, 362.0088).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


