1. Introduction
Gorgonian corals of the genus
Junceella (phylum, Cnidaria; class, Anthozoa; order, Gorgonacea; family, Ellisellidae) are widely distributed in the subtropical and tropical waters of the Indo-Pacific Ocean as whip-shaped unbranched colonies of variable colors. These animals are well-known as a source of highly oxidized diterpenes of the briarane class [
1,
2]. Briarane-related natural products are characterized by the presence of a γ-lactone fused to a bicyclo[8.4.0] ring system [
3] and have attracted great attention due to their chemical diversity and wide spectrum of bioactivities, including cytotoxic [
4], anti-inflammatory [
5], antibacterial [
6], immunomodulatory [
7], anti-fouling [
8] and insecticidal [
9] effects. Chemical investigation on the genus of
Junceella can be traced back to 1983, at which time the isolation of junceellin [
10] was reported from the Chinese gorgonian
Junceella squamata [
1]. Since then, more than 140 briarane diterpenes have been isolated from members of this genus, which involves four species, namely
J.
juncea,
J.
fragilis,
J.
gemmacea and
J.
squamata [
1,
11]. Previous research activities have been focused on
J.
juncea and
J.
fragilis, and only a few reports have dealt with corals belonging to the species
J.
gemmacea and
J.
squamata [
2].
In our ongoing search for novel and bioactive secondary metabolites from marine invertebrates of the South China Sea, we were attracted by the potential medicinal value of the briarane diterpenoids. Previously, we reported the isolation of a series of briarane-type diterpenes with antimicrobial and tumor cell growth inhibition activities from the gorgonian,
Dichotella gemmacea [
12,
13,
14,
15]. These results inspired us to continue to examine this class of metabolites, leading to the present investigation of the gorgonian
J. gemmacea which was previously reported to be a rich source of briarane diterpenes. Chemical investigation on the acetone extract of these animals resulted in the isolation of four new briarane diterpenes, junceellolides M–P (
1–
4), together with seven known ones, namely junceellolide A (
5), junceellin A (
6), praelolide (
7), juncin ZI (
8), junceellolide B (
9), junceellolide C (
10) and junceellolide D (
11) (
Figure 1). The structures of these compounds were elucidated by extensive spectroscopic analysis (
1H and
13C NMR, DEPT, HSQC, HMBC, NOESY,
1H–
1H COSY and HRESIMS) and comparison with the reported analytical data for the known compounds. We herein report the isolation, structural determination and bioactivities of these new compounds.
2. Results and Discussion
Colonies of the gorgonian coral
J. gemmacea (Valenciennes) were immediately frozen after collection and stored at −20 °C until extraction. The frozen organism was cut into small pieces and extracted exhaustively with acetone and methanol at room temperature. The Et
2O-soluble portion (13.0 g) of the acetone extract was chromatographed repeatedly over silica gel, Sephadex LH-20, and semi-preparative RP-HPLC, to afford compounds
1–
11. By extensive spectroscopic analysis combined with careful comparison with the reported data, the structures of the known compounds were determined as junceellolide A (
5) [
16], junceellin A (
6) [
1,
10], praelolide (
7) [
16], juncin ZI (
8) [
17], junceellolide B (
9) [
16], junceellolide C (
10) [
16] and junceellolide D (
11) [
16]. Junceellin A (
6) and juncin ZI (
8) were once reported from
J.
squamata and
J.
juncea, respectively, and their structures were elucidated on the basis of ESIMS, UV, IR and NMR techniques. The structures of junceellolides A–D (
5,
9–
11) and praelolide (
7), previously obtained from the South China Sea gorgonian,
J.
fragilis, were also established by extensive spectroscopic analysis.
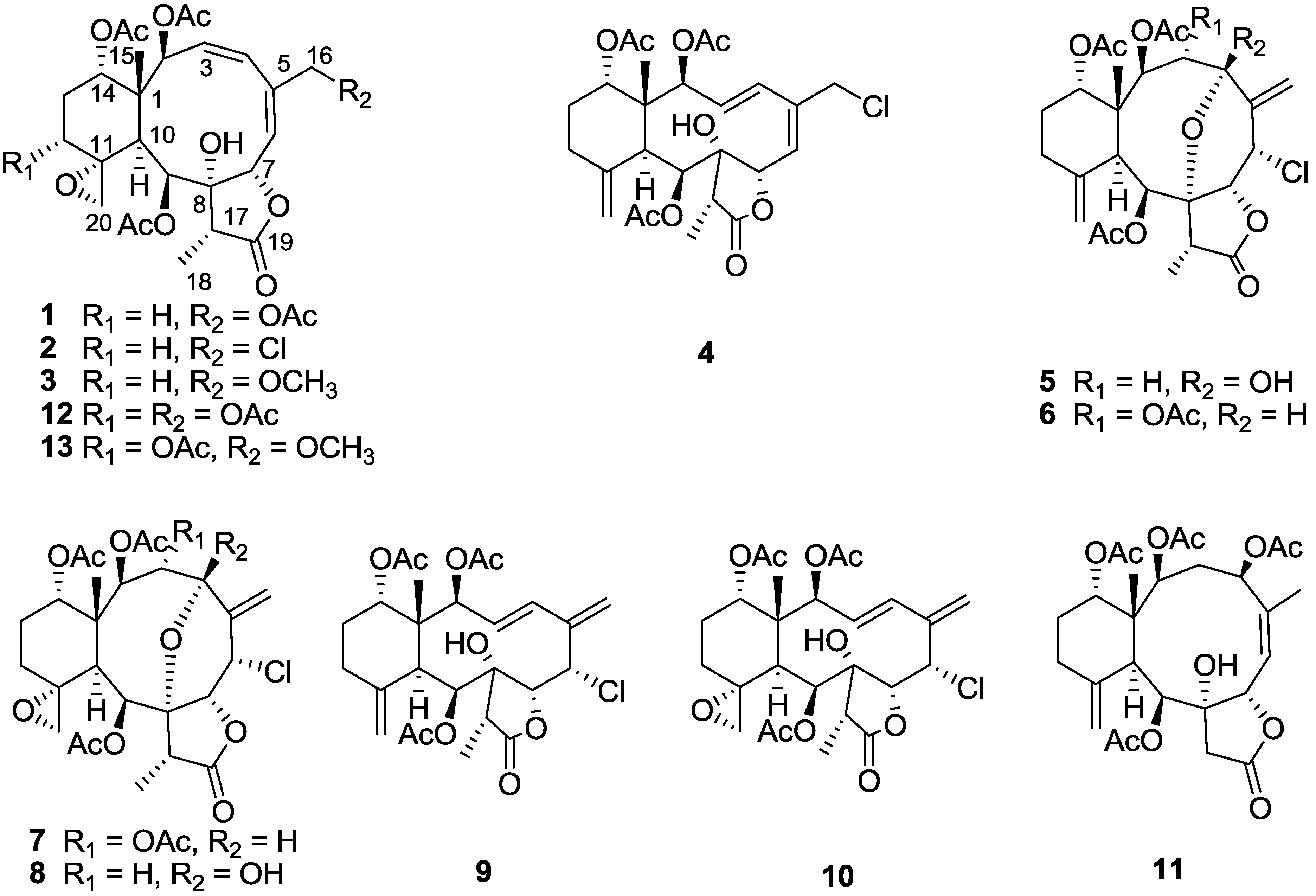
Figure 1.
Structures of compounds 1–11.
Figure 1.
Structures of compounds 1–11.
Junceellolide M (
1) was isolated as a white amorphous powder and had a molecular formula of C
28H
36O
12 deduced from HRESIMS, indicating eleven degrees of unsaturation. The IR spectrum of
1 showed absorption bands for γ-lactone (1781 cm
−1) and ester carbonyl (1736 cm
−1) groups. This observation was consistent with the signals in the
13C NMR and DEPT spectra (
Table 1) for nine sp
2 carbon atoms (5 × OC=O, CH=CH, CH=C) at lower field and nineteen sp
3 carbon atoms at higher field (1 × C, 2 × CH, 2 × CH
2, 6 × CH
3, 2 × OC, 4 × OCH, 2 × OCH
2), accounting for seven double-bond equivalents. The remaining double bond equivalents were due to the presence of three rings in the molecule.
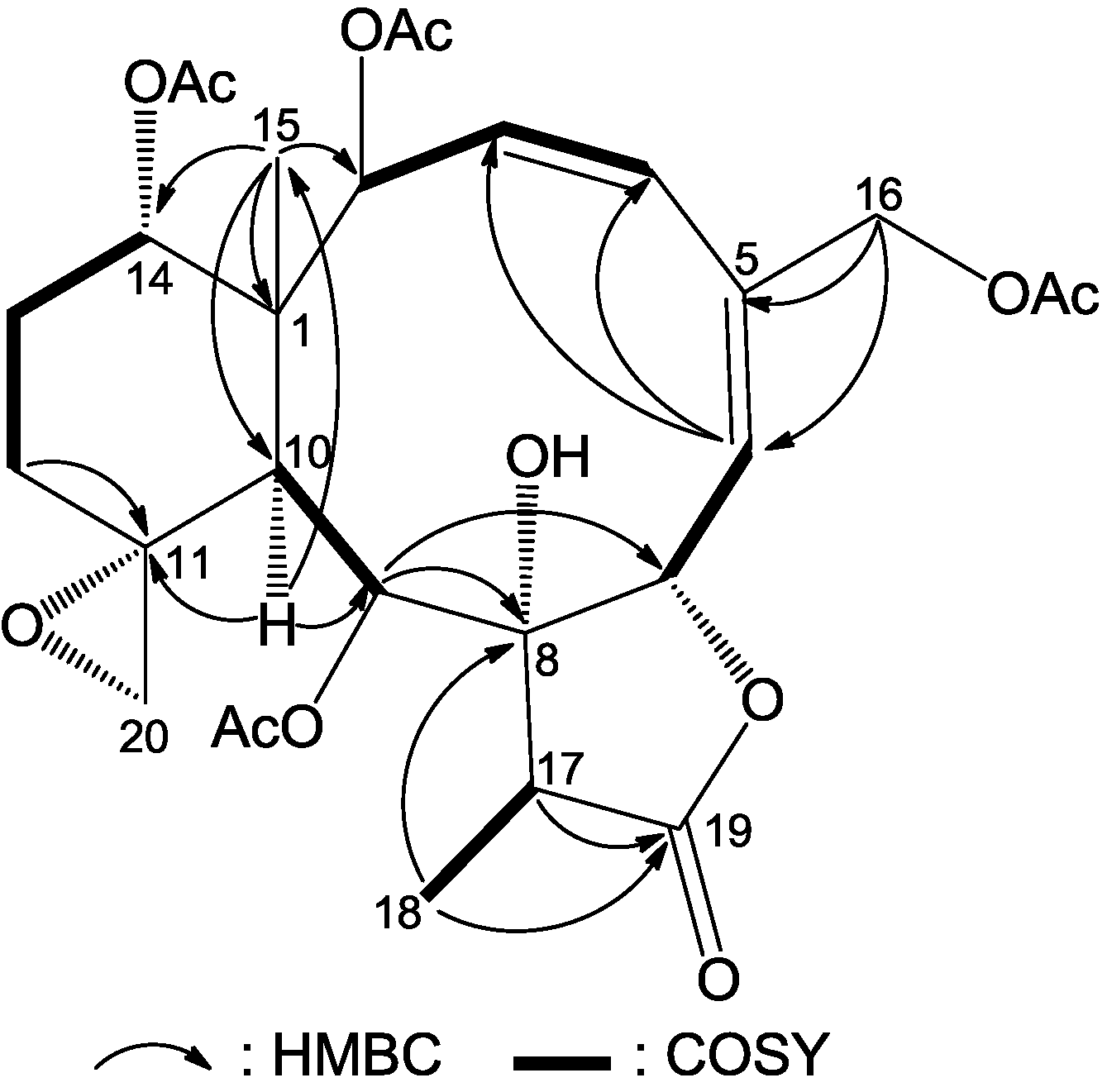
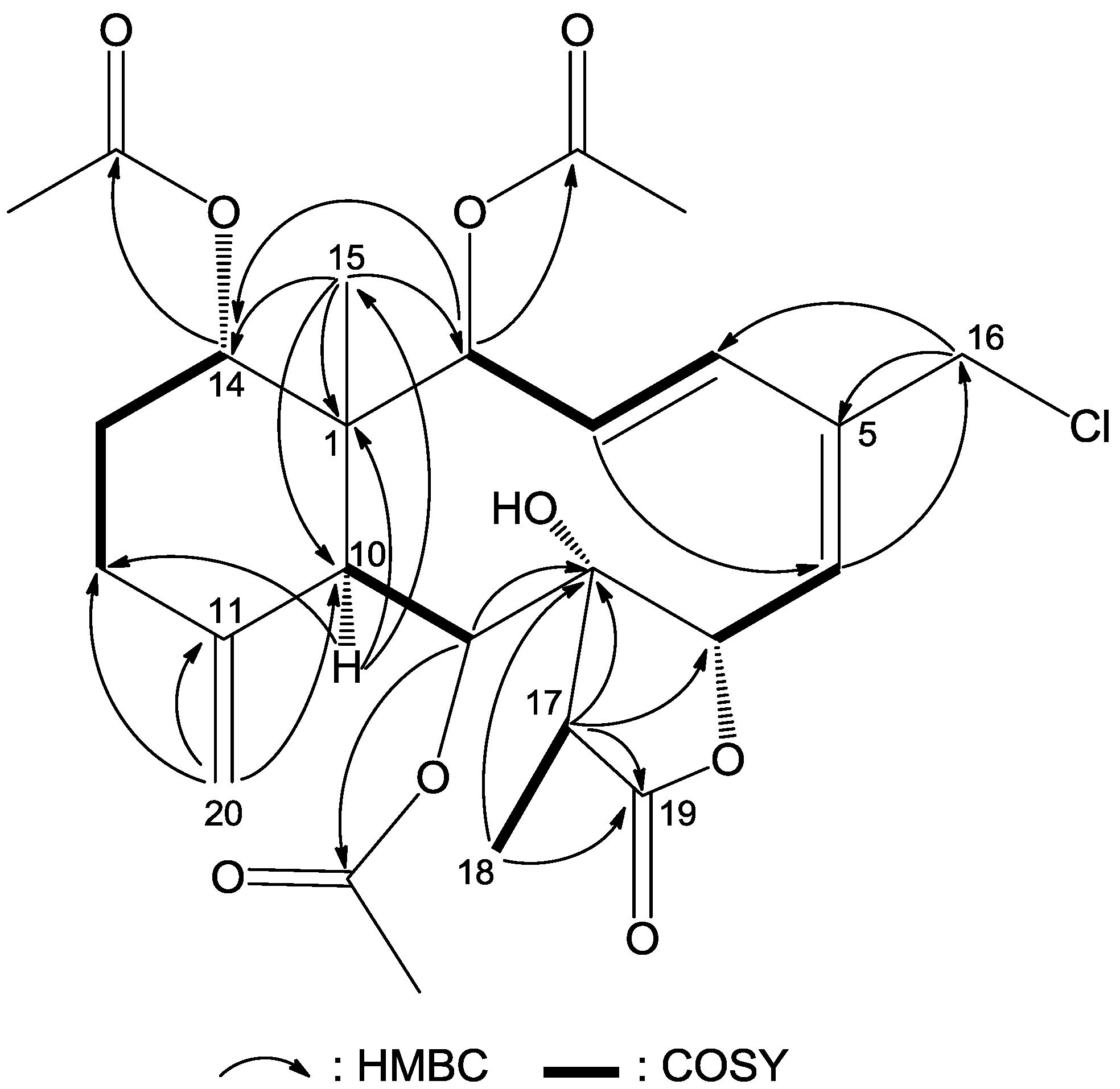
The NMR data for compound
1 were almost identical to those of frajunolide D (
12) [
18], an analogue isolated from the gorgonian
J. fragilis, except for the absence of one acetate ester group. Location of the missing acetate ester group to C-12 was indicated by the distinct correlations from H
2-12 to H
2-13 and to H-14 in a
1H–
1H COSY experiment of
1 (
Figure 2). The planar structure of
1 was further supported by
1H–
1H COSY and HMBC data, as shown in
Figure 2.
Table 1.
1H NMR data of compounds 1–4 (in CDCl3, J in Hz). s = singlet; d = doublet; t = triplet; m = multiplet; br s = broad singlet; br d = broad doublet; dd = doublet of doublets; tt = triplet of triplets; ov = overlapped signals.
Table 1.
1H NMR data of compounds 1–4 (in CDCl3, J in Hz). s = singlet; d = doublet; t = triplet; m = multiplet; br s = broad singlet; br d = broad doublet; dd = doublet of doublets; tt = triplet of triplets; ov = overlapped signals.
| Position | 1 a | 2 b | 3 a | 4 a |
|---|
| 2 | 5.61 ov | 5.52 d (9.5) | 5.60 ov | 5.61 ov |
| 3 | 5.59 ov | 5.61 dd (9.5, 10.5) | 5.59 ov | 6.16 dd (16.9, 5.7) |
| 4 | 6.26 d (9.1) | 6.33 d (10.5) | 6.26 d (9.1) | 6.41 d (16.9) |
| 6 | 5.76 d (8.6) | 6.01 d (9.0) | 5.87 d (8.3) | 5.62 ov |
| 7 | 4.97 d (8.6) | 4.95 dd (9.0, 1.5) | 4.98 d (8.3) | 5.69 br s |
| 9 | 4.75 ov | 4.75 d (5.5) | 4.74 d (5.0) | 5.94 d (5.7) |
| 10 | 3.12 d (5.1) | 3.10 d (5.5) | 3.13 d (5.0) | 3.28 d (5.7) |
| 12α | 2.20 ov | 2.20 ov | 2.20 ov | 2.22 ov |
| 12β | 1.10 ov | 1.10 ov | 1.10 ov | 2.25 ov |
| 13β | 1.73 m | 1.73 m | 1.72 ov | 1.78 ov |
| 13α | 1.96 ov | 1.94 ov | 1.93 ov | 1.95 ov |
| 14 | 4.90 br s | 4.89 br s | 4.88 br s | 4.88 br s |
| 15-Me | 1.00 s | 1.01 s | 1.00 s | 0.90 s |
| 16a | 5.31 d (16.0) | 4.62 d (13.0) | 4.43 d (14.6) | 4.16 ov |
| 16b | 4.72 d (16.0) | 4.56 d (13.0) | 4.27 d (14.6) | 4.17 ov |
| 17 | 2.27 q (7.0) | 2.27 q (6.7) | 2.26 q (7.0) | 2.64 q (7.2) |
| 18-Me | 1.14 d (7.0) | 1.13 d (6.7) | 1.14 d (7.0) | 1.25 d (7.2) |
| 20a | 3.50 br s | 3.50 br s | 3.51 br s | 5.11 s |
| 20b | 2.65 d (2.5) | 2.65 d (1.5) | 2.64 d (2.2) | 5.16 s |
| -OAc | 2.18 s | 2.18 s | 2.18 s | 2.25 s |
| | 2.14 s | 2.04 s | 2.08 s | 2.19 s |
| | 2.09 s | 1.99 s | 1.98 s | 2.10 s |
| | 1.97 s | | | |
| -OH | 2.85 d (1.7) | 2.83 d (1.5) | | |
| OMe | | | 3.43 s (3H) | |
Figure 2.
Key HMBC (arrow H→C) and 1H–1H COSY (bond) correlations for compound 1.
Figure 2.
Key HMBC (arrow H→C) and 1H–1H COSY (bond) correlations for compound 1.
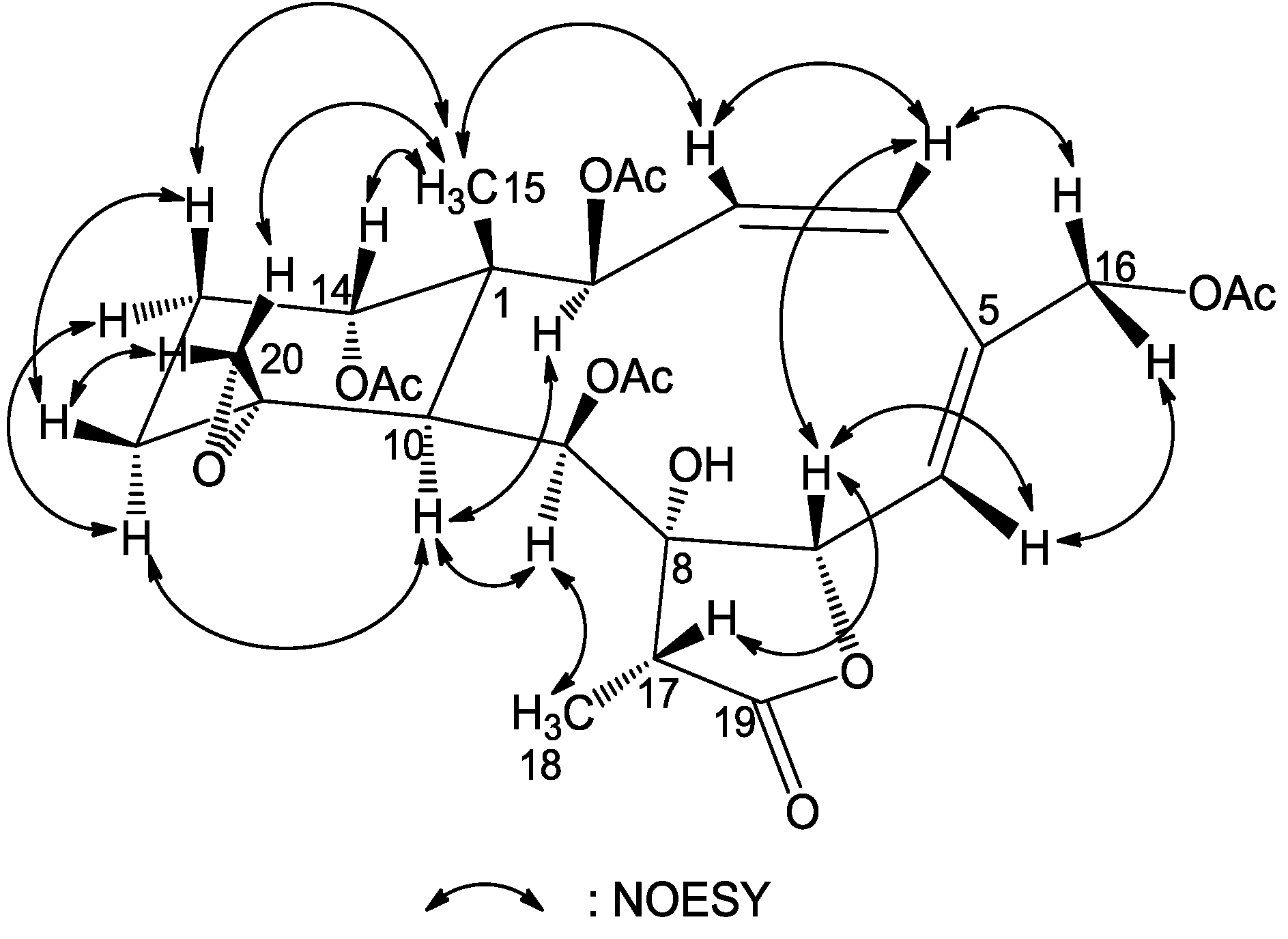
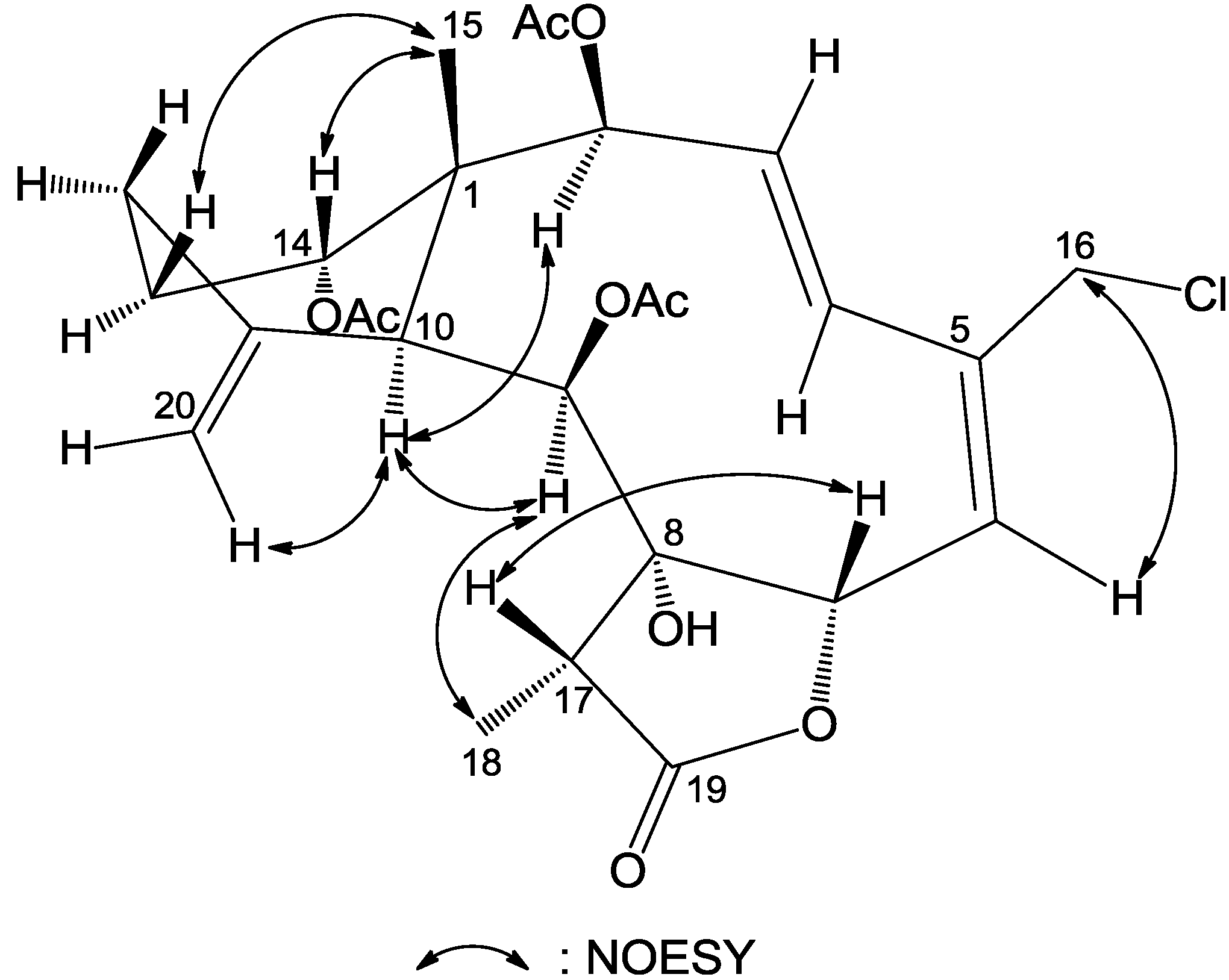
The relative configuration of
1 was proven to be the same as that of frajunolide D (
12) by a NOESY experiment (
Figure 3), in which we have arbitrarily chosen the β
-orientation of H-13β, H-14, Me-15, H-17 and H
2-20 and an α-orientation of H-2, H-9, H-10, Me-18 and 8-OH. The geometry of the Δ
3 double bond was assigned as
Z based on the proton coupling constant between H-3 and H-4 (
J = 8.7 Hz), while the geometry of the Δ
5 double bond was
E as deduced from an NOE correlation between H-6 and H-16a. The chemical shift value of C-11 and C-20 (δ
C 60.1 and 50.5, respectively) supported the α-configurational assignment of the 11,20-epoxy group. It has been reported that the
13C NMR shifts for C-11 and C-20 are at δ 62–63 and 58–60 when the epoxy group is β
-oriented and at δ 55–61 and 47–52 ppm when α-oriented [
19]. The relative configuration of
1 was thus determined as (1
R*,2
S*,3
Z,5
E,7
S*,8
S*,9
S*,10
S*,11
R*,14
S*,17
R*).
Figure 3.
Key NOESY correlations for compound 1.
Figure 3.
Key NOESY correlations for compound 1.
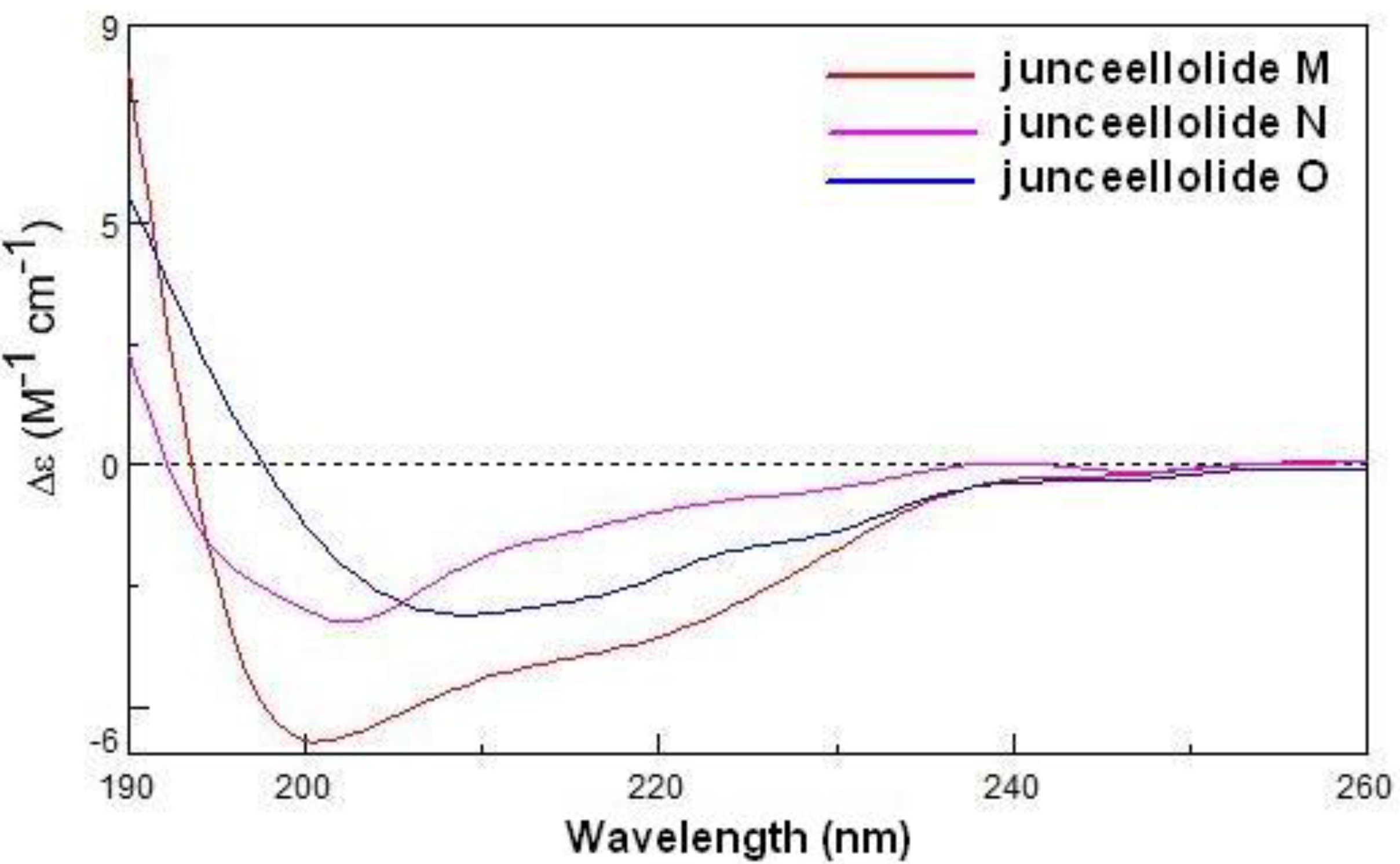
Because compound
1 contained the same lactone and diene chromophores as gemmacolide N (
13) [
14], and because they differed only in the nature of the ester group at the C-12 and C-16 positions, the ECD spectrum of gemmacolide N could be used as an ECD reference for the configurational assignment of junceellolide M. Based on a negative ECD transition in the region 250–200 nm and a positive band below 200 nm, the absolute configuration of
1 was determined as (–)-(1
R,2
S,3
Z,5
E,7
S,8
S,9
S,10
S,11
R,14
S,17
R) (
Figure 4).
Figure 4.
The ECD spectra of compounds 1–3 in acetonitrile.
Figure 4.
The ECD spectra of compounds 1–3 in acetonitrile.
Junceellolide N (
2) was obtained as a white powder. The molecular formula was determined to be C
26H
33O
10Cl by HRESIMS. An isotopic ratio of 3:1 was observed in the molecular ion peaks at
m/z 563.1663/565.1782 [M + Na]
+, further indicating the presence of one chlorine atom. The comparison of the overall
1H and
13C NMR data of
2 with those of
1 (
Table 1) revealed a close similarity, except that the acetyl group at C-16 in
1 was replaced by a chlorine atom in
2. The structure was fully supported by the 2D NMR experiments, including
1H–
1H COSY, HMBC and NOESY. Compound
2 also gave a similar negative ECD band at 202 nm and a positive one below 200 nm (
Figure 4), and thus its absolute configuration was then determined as (−)-(1
S,2
S,3
Z,5
E,7
S,8
S,9
S,10
S,11
R,14
S,17
R).
Junceellolide O (3) had a molecular formula of C27H36O11, as deduced from the HRESIMS. The 1H and 13C NMR data of 3 also closely resembled those of 1, except for the replacement of the acetyl group at C-16 in 1 by a methoxy group (δH 3.43, δC 58.5) in 3. This assignment was further confirmed by the distinct HMBC correlation from H2-16 to the methoxy carbon (δC 58.5). The relative and absolute configuration of 3 was proven to be the same as that of 1 by analysis of the NOESY and ECD spectra, and was thus determined as (+)-(1S,2S,3Z,5E,7S,8S,9S,10S,11R,14S,17R).
Junceellolide P (
4) had a molecular formula of C
26H
33O
9Cl, as determined by HRESIMS, and showed the presence of a chlorine atom in the molecule by the isotopic ratio of molecular ion peaks
m/z 547.1715/549.1753 [M + Na]
+ (3:1). Its IR spectrum indicated the presence of hydroxy (3497 cm
−1), γ-lactone (1777 cm
−1) and ester (1740 cm
−1) functionalities. The
1H and
13C NMR spectra of
4 demonstrated a set of typical NMR signals for a briarane diterpenoid (
Table 1), including ten sp
2 carbon atoms (4 × OC=O, CH=CH, CH=C, CH
2=C) at lower field and sixteen sp
3 carbon atoms at higher field (1 × C, 2 × CH, 2 × CH
2, 5 × CH
3, 1 × OC, 4 × OCH, 1 × CH
2Cl).
The gross structure of compound
4 was characterized by a detailed analysis of 2D NMR spectra. The
1H–
1H COSY spectrum gave five proton spin systems of H-2/H-3/H-4, H-6/H-7, H-9/H-10, H
2-12/H
2-13/H-14 and H-17/H
3-18. The connections of these proton sequences led to the establishment of the planar structure of
4 by the observation of distinct HMBC correlations from H
3-15 to C-1, C-2, C-10 and C-14, H
2-16 to C-4 and C-5, H-6 to C-16, H-17 to C-7, C-8, C-9 and C-19, H-9 to C-8 and H
2-20 to C-10, C-11 and C-12, respectively (
Figure 5). The relative configurations at chiral centers were proven to be the same as those of compounds
1–
3. Interestingly, the large coupling constant for the olefinic protons (
J = 16.8 Hz) indicated an
E geometry of Δ
3 in
4 in contrast to those of the
Z double bonds in structures
1–
3. To our knowledge, this is the first report of a
trans Δ
3,5 conjugated diene in a briarane diterpenoid. The relative configuration of
4 was established by NOE (
Figure 6) and coupling constant analysis whereas we propose its absolute configuration as (1
R,2
S,3
E,5
E,7
S,8
S,9
S,10
S,11
R,14
S,17
R)-
4, due to its biogenetic correlation with those of compounds
1–
3.
Compounds
1–
11 were evaluated for their ability to inhibit tumor cell growth using the A549, MG63 and SMMC-7721 cell lines [
20]. None of these compounds exhibited growth inhibitory effects against these cell lines in the
in vitro bioscreening (IC
50 ≥ 40 µM).
Figure 5.
Key HMBC correlations (arrow H→C) and COSY (bond) spin coupling systems for compound 4.
Figure 5.
Key HMBC correlations (arrow H→C) and COSY (bond) spin coupling systems for compound 4.
Figure 6.
Key NOESY correlations of compound 4.
Figure 6.
Key NOESY correlations of compound 4.
3. Experimental Section
3.1. General Experimental Procedures
Commercial silica gel (Yantai, China, 200–300; 400–500 mesh) and RP silica gel (Merck, Darmstadt, Germany, 43–60 µm) were used for column chromatography (CC). Sephadex LH-20 (GE Healthcare Bio-Sciences AB, SE-751 84 Uppsala, Sweden) was used for either purification or separation. TLC was carried out on precoated silica gel plates (Yantai, China, HSGF-254) and RP silica gel (Macherey-Nagel, Düren, Germany, RP-18 F254), and spots were detected on TLC under UV and visualized by spraying with anisaldehyde-sulfuric acid reagent, followed by heating. HPLC was performed using a system comprised of an Agilent G1311A pump, an Agilent G1315B DAD detector and a Rheodyne 7725 injection port. A semi-preparative normal-phase column (YMC Pack ODS-A, 250 × 10 mm I.D., particle size 5 µm, 250 × 10 mm, YMC Europe GmbH, Dinslaken, Germany) was used for HPLC. The NMR data were recorded on a Bruker DRX 400 and Avance 500 spectrometers at 400 or 500 MHz for 1H and 100 or 125 MHz for 13C, respectively. Chemical shifts are reported in parts per million (δ), with the use of the residual CDCl3 signal (δH = 7.27 ppm) as an internal standard for 1H NMR and CDCl3 (δC = 77.02 ppm) for 13C NMR, and the coupling constants (J) were in Hz. The 1H and 13C NMR assignments were supported by 1H–1H COSY, HSQC, HMBC and NOESY experiments. The following abbreviations are used to describe spin multiplicity: s denotes singlet; d denotes doublet; t denotes triplet; m denotes multiplet; br s denotes broad singlet; br d denotes broad doublet; dd denotes doublet of doublets; tt denotes triplet of triplets; ov denotes overlapped signals. Optical rotations were measured in CH2Cl2 with an Autopol IV polarimeter at the sodium D line (590 nm). Infrared spectra were recorded in thin polymer films on a Nexus 470 FT-IR spectrophotometer (Nicolet, USA); peaks are reported in cm−1. UV absorption spectra were recorded on a Varian Cary 100 UV-Vis spectrophotometer; peak wavelengths are reported in nanometers. Circular dichroism spectra were recorded with a JASCO J-715 circular dichroism spectropolarimeter. The HRMS were performed on a Q-TOF micro-mass spectrometer (resolution: 5000). An isopropyl alcohol solution of sodium iodide (2 mg/mL) was used as a reference compound.
3.2. Animal Material
Specimens of the gorgonian coral Junceella gemmacea were collected from the South China Sea in October 2011, and identified by Xiu-Bao Li of the South China Sea Institute of Oceanology, Academia Sinica. The voucher specimen was deposited in the Second Military Medical University, Shanghai, China.
3.3. Extraction and Isolation
The frozn animals (2500 g, wet weight) were cut into small pieces and extracted ultrasonically for four times (3000 mL × 4) with acetone and MeOH at room temperature. The organic extracts were concentrated under vacuum to give a residue, which was suspended into H2O and extracted with diethyl ether and n-butanol for four times, respectively. The Et2O-soluble portion was separated on a Sephadex LH-20 column (CH2Cl2:MeOH 1:1) giving fractions 1–10 (Fr.1~Fr.10). Fr.6 was subjected to SiO2 column chromatography using solvents of increasing polarity from petroleum to acetone (TLC (GF 254) monitoring) to obtain eleven subfractions (P.1~P.11). P.11 was separated by reversed-phase silica gel chromatography (gradient MeOH/H2O, from 30:70 to 0:100), followed by semi-preparative RP-HPLC (MeOH/H2O, 58:42, flow rate of 2.0 mL/min), to yield compounds 1 (4.2 mg, tR 35 min) and 3 (3.7 mg, tR 32 min). Fr.8 was fractioned on silica gel eluting with petroleum/acetone (stepwise, 20:1–0:1) and then chromatographed on a gravity column with ODS using MeOH/water (gradient from 30:70 to 0:100, in 10% increments) as the eluent to afford subfractions S.10 and S.11. S.10 was isolated by reversed-phase silica gel chromatography (gradient elution from MeOH/H2O (30:70) to 100% MeOH, in 10% increments) and successively subjected to RP-HPLC (MeOH/H2O 79:21, 2 mL/min), producing 5 (2.6 mg, tR 29 min). S.11 was chromatographed over RP-silica gel column chromatography using a gradient of MeOH/H2O (30:70 to 0:100, in 10% increments) to yield subfractions R.4 and R.5. A HPLC chromatographic isolation on R.4 (MeOH/H2O 62:38, 2 mL/min) led to the preparation of 2 (2.4 mg, tR 60 min), 8 (2.3 mg, tR 39 min), 10 (4.9 mg, tR 53 min) and 11 (3.4 mg, tR 83 min). Furthermore, R.5 was further separated by HPLC using MeOH/H2O (56:44) at the flow rate of 2.0 mL/min and provided 4 (5.9 mg, tR 51 min) and 9 (1.6 mg, tR 54 min). Using petroleum/acetone (7:1) as the eluent, the crystal precipitated from Fr.8 furnished 6 (35.0 mg) and 7 (16.9 mg) on Silica gel column chromatography.
Junceellolide M (1): white amorphous powder;
![Marinedrugs 12 00589 i001]()
= −0.85° (
c 0.47, CH
2Cl
2); UV (MeOH) 230 nm; CD (CH
3CN,
c 3.0 × 10
−4) λ
max (Δε) positive below 193.5 nm, 200 (−5.67) nm; IR (film) ν
max 2959, 2930, 1781, 1736, 1252, 1225 cm
−1;
1H NMR spectroscopic data, see
Table 1;
13C NMR spectroscopic data, see
Table 2; HRESIMS
m/z 587.2100 [M + Na]
+, calcd. for C
28H
36O
12Na, 587.2104.
Table 2.
13C NMR data of compounds 1–4 (in CDCl3, J in Hz).
Table 2.
13C NMR data of compounds 1–4 (in CDCl3, J in Hz).
| Position | 1 a | 2 b | 3 a | 4 a |
|---|
| 1 | 47.1, C | 47.2, C | 47.3, C | 49.1, C |
| 2 | 74.7, CH | 74.6, CH | 74.8, CH | 75.5, CH |
| 3 | 132.6, CH | 132.3, CH | 131.7, CH | 137.8, CH |
| 4 | 127.2, CH | 127.8, CH | 128.2, CH | 126.4, CH |
| 5 | 139.6, C | 140.0, C | 141.4, C | 140.0, C |
| 6 | 122.5, CH | 126.2, CH | 123.0, CH | 127.6, CH |
| 7 | 78.8, CH | 78.6, CH | 79.0, CH | 80.7, CH |
| 8 | 80.9, C | 80.9, C | 80.9, C | 80.7, C |
| 9 | 64.6, CH | 64.6, CH | 64.7, CH | 73.4, CH |
| 10 | 37.8, CH | 37.8, CH | 37.8, CH | 43.2, CH |
| 11 | 60.1, C | 60.0, C | 60.1, C | 148.9, C |
| 12 | 29.2, CH2 | 29.2, CH2 | 29.3, CH2 | 31.2, CH2 |
| 13 | 25.1, CH2 | 25.0, CH2 | 25.1, CH2 | 27.8, CH2 |
| 14 | 74.4, CH | 74.5, CH | 74.7, CH | 78.8, CH |
| 15 | 14.4, CH3 | 14.4, CH3 | 14.5, CH3 | 15.7, CH3 |
| 16 | 63.0, CH2 | 44.9, CH2 | 72.4, CH2 | 47.5, CH2 |
| 17 | 44.0, CH | 44.0, CH | 44.1, CH | 45.4, CH |
| 18 | 6.4, CH3 | 6.4, CH3 | 6.4, CH3 | 8.2, CH3 |
| 19 | 175.6, C | 175.6, C | 175.7, C | 175.4, C |
| 20 | 50.5, CH2 | 50.5, CH2 | 50.5, CH2 | 111.4, CH2, |
| -OAc | 170.3, C | 170.2, C | 170.3, C | 170.4, C |
| | 21.6, CH3 | 21.6, CH3 | 21.6, CH3 | 21.5, CH3 |
| | 170.2, C | 170.2, C | 170.2, C | 169.7, C |
| | 20.9, CH3 | 21.3, CH3 | 21.3, CH3 | 21.7, CH3 |
| | 170.4, C | 169.7, C | 169.5, C | 169.9, C |
| | 21.2, CH3 | 21.1, CH3 | 21.2, CH3 | 20.9, CH3 |
| | 169.4, C | | | |
| | 21.1, CH3 | | | |
| OMe | | | 58.5, CH3 | |
Junceellolide N (2): white amorphous powder;
![Marinedrugs 12 00589 i001]()
= −10.89° (
c 0.32, CH
2Cl
2); UV (MeOH) 418, 231 nm; CD (CH
3CN,
c 3.0 × 10
−4) λ
max (Δε) positive below 192 nm, 202.5 (−3.23) nm; IR (film) ν
max 2959, 2930, 1782, 1731, 1288, 1274 cm
−1;
1H NMR spectroscopic data, see
Table 1;
13C NMR spectroscopic data, see
Table 2; HRESIMS
m/z 563.1663 [M + Na]
+, calcd. for C
26H
33O
10ClNa, 563.1660.
Junceellolide O (3): white amorphous powder;
![Marinedrugs 12 00589 i001]()
= +8.75° (
c 0.04, CH
2Cl
2); UV (MeOH) 228 nm; CD (CH
3CN,
c 3.0 × 10
−4) λ
max (Δε) positive below 198 nm, 209 (−3.08) nm; IR (film) ν
max 2959, 2930, 1780, 1732, 1272 cm
−1;
1H NMR spectroscopic data, see
Table 1;
13C NMR spectroscopic data, see
Table 2; HRESIMS
m/z 559.2158 [M + Na]
+, calcd. for C
27H
36O
11Na, 559.2155.
Junceellolide P (4): white acicular crystal,
![Marinedrugs 12 00589 i001]()
= −59.29° (
c 0.28, CH
2Cl
2); UV (MeOH) 229 nm; IR (film) ν
max 3456, 1777, 1740, 1236 cm
−1;
1H NMR spectroscopic data, see
Table 1;
13C NMR spectroscopic data, see
Table 2; HRESIMS
m/z 547.1715 [M + Na]
+, calcd. for C
26H
33O
9ClNa, 547.1711.
3.4. Cytotoxicity Assay
Compounds
1–
11 were evaluated for cytotoxicity against human lung adenocarcinoma (A549), human osteosarcoma cell (MG63) and human hepatocellular carcinoma cell lines (SMMC-7721), using a modification of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric method [
21]. Adriamycin was used as a positive control.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





 = −0.85° (c 0.47, CH2Cl2); UV (MeOH) 230 nm; CD (CH3CN, c 3.0 × 10−4) λmax (Δε) positive below 193.5 nm, 200 (−5.67) nm; IR (film) νmax 2959, 2930, 1781, 1736, 1252, 1225 cm−1; 1H NMR spectroscopic data, see Table 1; 13C NMR spectroscopic data, see Table 2; HRESIMS m/z 587.2100 [M + Na]+, calcd. for C28H36O12Na, 587.2104.
= −0.85° (c 0.47, CH2Cl2); UV (MeOH) 230 nm; CD (CH3CN, c 3.0 × 10−4) λmax (Δε) positive below 193.5 nm, 200 (−5.67) nm; IR (film) νmax 2959, 2930, 1781, 1736, 1252, 1225 cm−1; 1H NMR spectroscopic data, see Table 1; 13C NMR spectroscopic data, see Table 2; HRESIMS m/z 587.2100 [M + Na]+, calcd. for C28H36O12Na, 587.2104.