Recognition of LPS by TLR4: Potential for Anti-Inflammatory Therapies

Abstract
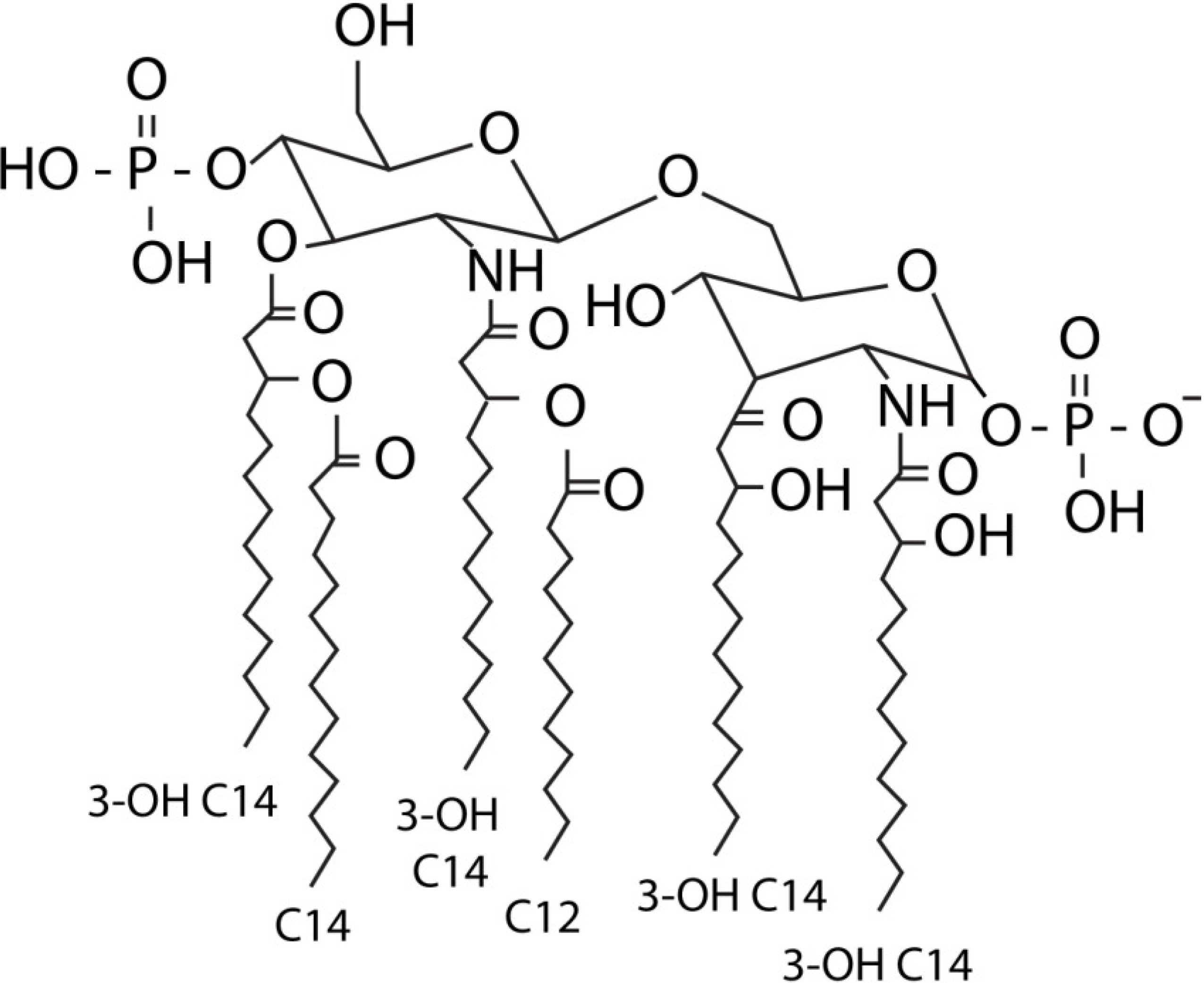
:1. LPS

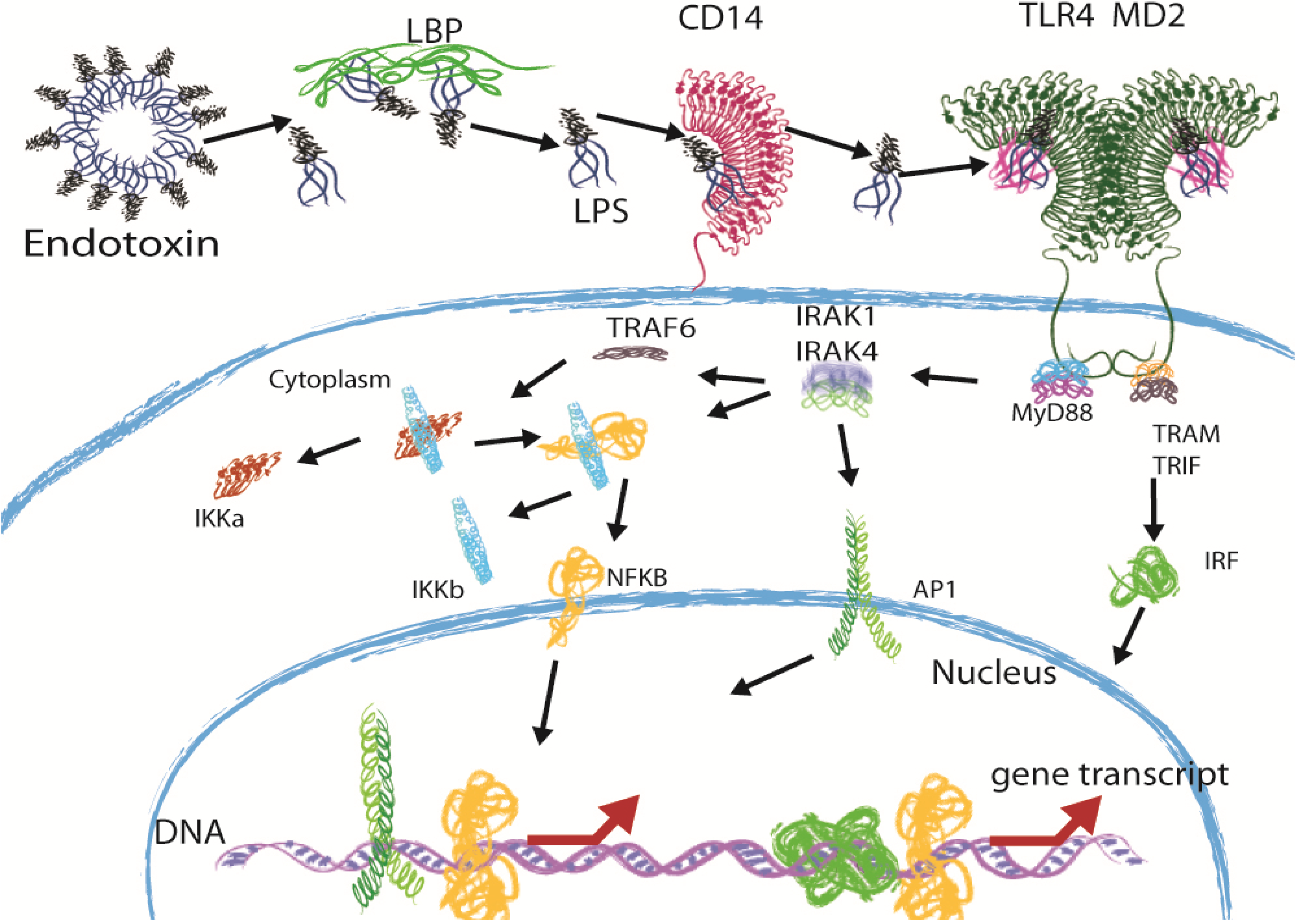
2. Immune Recognition of LPS through the TLR4 Pathway

3. Sepsis and the Potential of Structurally Different LPS Molecules as Antagonists
4. Structure and Immune Recognition of E. coli Lipid A
5. Immune Recognition of Lipid A Structures of Other Terrestrial Bacteria
6. Decreased Lipid A Acylation Reduces Immune Potency
7. Immune Recognition of Lipid A Structures from Marine Bacteria

{kind=link}
{kind=link}
| Species | No. of Acyl Chains | No. of Phosphorylations | Reference |
|---|---|---|---|
| Pseudoalteromonas haloplanktis (TAC 125) | 5 | 2 | [30] |
| Pseudoalteromonas haloplanktis (ATCC 14393) | 5 | 2 | [1] |
| Alteromonas addita | 5 | 2 | [31] |
| Marinomonas vaga | 5 | 1 | [32] |
| Pseudoalteromonas issachenkonii | 4 (5) | 2 | [33] |
| Alteromonas macleodii | 4 (5) | 2 | [34] |
| Synechococcus strains CC9311 and WH8102 | 4 | 0 | [35] |
| Shewanella pacifica | 6 | 2 | [36] |
| Chryseobacterium scophtalmum | 2 | 1 | [1] |
| Marinomonas communis | 5 | 1 | [15] |
| Marinomonas mediterranea | 5 (6) | 2 | [15] |
8. Main Differences between Marine and Terrestrial Lipid A Structures
9. Why Do Marine Bacteria Have Different Lipid A?
10. Drugs Based on Modified LPS in Sepsis Therapy Have Limited Success
11. Bacterial TLR4 Inhibitors as Therapeutics against Sepsis
12. Compounds that Target the TLR4 Signaling Pathway
13. Future Inhibitors of the TLR4 Signaling Pathway
14. Concluding Remarks
Conflicts of Interest
References
- Leone, S.; Silipo, A.; Nazarenko, E.L.; Lanzetta, R.; Parrilli, M.; Molinaro, A. Molecular structure of endotoxins from Gram-negative marine bacteria: An update. Mar. Drugs 2007, 5, 85–112. [Google Scholar] [CrossRef]
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Mamat, U.; Schmidt, G.; Loppnow, H.; Ulmer, A.J.; Zähringer, U.; Seydel, U.; di Padova, F. Bacterial endotoxin: Molecular relationships of structure to activity and function. FASEB J. 1994, 8, 217–225. [Google Scholar]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef]
- Van Amersfoort, E.S.; van Berkel, T.J.; Kuiper, J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin. Microbiol. Rev. 2003, 16, 379–414. [Google Scholar] [CrossRef]
- Solov’eva, T.; Davydova, V.; Krasikova, I.; Yermak, I. Marine compounds with therapeutic potential in Gram-negative sepsis. Mar. Drugs 2013, 11, 2216–2229. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Golenbock, D.T. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes Infect./Inst. Pasteur 2004, 6, 1361–1367. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.-Y.; Huffel, C.V.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective lps signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in TLR4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Gioannini, T.L.; Teghanemt, A.; Zhang, D.; Coussens, N.P.; Dockstader, W.; Ramaswamy, S.; Weiss, J.P. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc. Natl. Acad. Sci. USA 2004, 101, 4186–4191. [Google Scholar] [CrossRef]
- Da Silva Correia, J.; Soldau, K.; Christen, U.; Tobias, P.S.; Ulevitch, R.J. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex: Transfer from CD14 to TLR4 and MD-2. J. Biol. Chem. 2001, 276, 21129–21135. [Google Scholar]
- DeMarco, M.L.; Woods, R.J. From agonist to antagonist: Structure and dynamics of innate immune glycoprotein MD-2 upon recognition of variably acylated bacterial endotoxins. Mol. Immunol. 2011, 49, 124–133. [Google Scholar] [CrossRef]
- Huber, M.; Kalis, C.; Keck, S.; Jiang, Z.; Georgel, P.; Du, X.; Shamel, L.; Sovath, S.; Mudd, S.; Beutler, B.; et al. R-form LPS, the master key to the activation ofTLR4/MD-2-positive cells. Eur. J. Immunol. 2006, 36, 701–711. [Google Scholar] [CrossRef]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. Tram couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef]
- Tanimura, N.; Saitoh, S.; Matsumoto, F.; Akashi-Takamura, S.; Miyake, K. Roles for LPS-dependent interaction and relocation of TLR4 and tram in TRIF-signaling. Biochem. Biophys. Res. Commun. 2008, 368, 94–99. [Google Scholar] [CrossRef]
- Raetz, C.R.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid a modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef]
- Vorobeva, E.V.; Krasikova, I.N.; Solov’eva, T.F. Influence of lipopolysaccharides and lipids a from some marine bacteria on spontaneous and Escherichia coli LPS-induced TNF-alpha release from peripheral human blood cells. Biochem. Biokhimiia 2006, 71, 759–766. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Schromm, A.B.; Brandenburg, K.; Loppnow, H.; Moran, A.P.; Koch, M.H.; Rietschel, E.T.; Seydel, U. Biological activities of lipopolysaccharides are determined by the shape of their lipid a portion. Eur. J. Biochem. 2000, 267, 2008–2013. [Google Scholar] [CrossRef]
- Erridge, C.; Moncayo-Nieto, O.L.; Morgan, R.; Young, M.; Poxton, I.R. Acinetobacter baumannii lipopolysaccharides are potent stimulators of human monocyte activation via Toll-like receptor 4 signalling. J. Med. Microbiol. 2007, 56, 165–171. [Google Scholar] [CrossRef]
- Leone, S.; Sturiale, L.; Pessione, E.; Mazzoli, R.; Giunta, C.; Lanzetta, R.; Garozzo, D.; Molinaro, A.; Parrilli, M. Detailed characterization of the lipid a fraction from the nonpathogen Acinetobacter radioresistens strain s13. J. Lipid Res. 2007, 48, 1045–1051. [Google Scholar] [CrossRef]
- Pelletier, M.R.; Casella, L.G.; Jones, J.W.; Adams, M.D.; Zurawski, D.V.; Hazlett, K.R.; Doi, Y.; Ernst, R.K. Unique structural modifications are present in the lipopolysaccharide from colistin-resistant strains of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2013, 57, 4831–4840. [Google Scholar] [CrossRef]
- Zahringer, U.; Lindner, B.; Inamura, S.; Heine, H.; Alexander, C. Tlr2—Promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 2008, 213, 205–224. [Google Scholar] [CrossRef]
- Girard, R.; Pedron, T.; Uematsu, S.; Balloy, V.; Chignard, M.; Akira, S.; Chaby, R. Lipopolysaccharides from legionella and rhizobium stimulate mouse bone marrow granulocytes via Toll-like receptor 2. J. Cell Sci. 2003, 116, 293–302. [Google Scholar] [CrossRef]
- Werts, C.; Tapping, R.I.; Mathison, J.C.; Chuang, T.H.; Kravchenko, V.; Saint Girons, I.; Haake, D.A.; Godowski, P.J.; Hayashi, F.; Ozinsky, A.; et al. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat. Immunol. 2001, 2, 346–352. [Google Scholar] [CrossRef]
- Hirschfeld, M.; Ma, Y.; Weis, J.H.; Vogel, S.N.; Weis, J.J. Cutting edge: Repurification of lipopolysaccharide eliminates signaling through both human and murine Toll-like receptor 2. J. Immunol. 2000, 165, 618–622. [Google Scholar] [CrossRef]
- Hellman, J.; Tehan, M.M.; Shaw Warren, H. Murein lipoprotein, peptidoglycan-associated lipoprotein, and outer membrane protein a are present in purified rough and smooth lipopolysaccharides. J. Infect. Dis. 2003, 188, 286–289. [Google Scholar] [CrossRef]
- Lee, H.-K.; Lee, J.; Tobias, P.S. Two lipoproteins extracted from Escherichia coli K-12 LCD25 lipopolysaccharide are the major components responsible for Toll-like receptor 2-mediated signaling. J. Immunol. 2002, 168, 4012–4017. [Google Scholar] [CrossRef]
- Kawahara, K.; Tsukano, H.; Watanabe, H.; Lindner, B.; Matsuura, M. Modification of the structure and activity of lipid a in Yersinia pestis lipopolysaccharide by growth temperature. Infect. Immun. 2002, 70, 4092–4098. [Google Scholar] [CrossRef]
- Hajjar, A.M.; Ernst, R.K.; Tsai, J.H.; Wilson, C.B.; Miller, S.I. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat. Immunol. 2002, 3, 354–359. [Google Scholar] [CrossRef]
- Stover, A.G.; da Silva Correia, J.; Evans, J.T.; Cluff, C.W.; Elliott, M.W.; Jeffery, E.W.; Johnson, D.A.; Lacy, M.J.; Baldridge, J.R.; Probst, P.; et al. Structure-activity relationship of synthetic Toll-like receptor 4 agonists. J. Biol. Chem. 2004, 279, 4440–4449. [Google Scholar]
- Corsaro, M.M.; Piaz, F.D.; Lanzetta, R.; Parrilli, M. Lipid a structure of Pseudoalteromonas haloplanktis TAC 125: Use of electrospray ionization tandem mass spectrometry for the determination of fatty acid distribution. J. Mass Spectrom. 2002, 37, 481–488. [Google Scholar] [CrossRef]
- Leone, S.; Molinaro, A.; Sturiale, L.; Garozzo, D.; Nazarenko, E.L.; Gorshkova, R.P.; Ivanova, E.P.; Shevchenko, L.S.; Lanzetta, R.; Parrilli, M. The outer membrane of the marine Gram-negative bacterium Alteromonas addita is composed of a very short-chain lipopolysaccharide with a high negative charge density. Eur. J. Org. Chem. 2007, 2007, 1113–1122. [Google Scholar] [CrossRef]
- Krasikova, I.N.; Kapustina, N.V.; Isakov, V.V.; Dmitrenok, A.S.; Dmitrenok, P.S.; Gorshkova, N.M.; Solov’eva, T.F. Detailed structure of lipid a isolated from lipopolysaccharide from the marine Proteobacterium marinomonas vaga ATCC 27119. Eur. J. Biochem. 2004, 271, 2895–2904. [Google Scholar] [CrossRef]
- Silipo, A.; Leone, S.; Lanzetta, R.; Parrilli, M.; Sturiale, L.; Garozzo, D.; Nazarenko, E.L.; Gorshkova, R.P.; Ivanova, E.P.; Gorshkova, N.M.; et al. The complete structure of the lipooligosaccharide from the halophilic bacterium Pseudoalteromonas issachenkonii KMM 3549 T. Carbohydr. Res. 2004, 339, 1985–1993. [Google Scholar] [CrossRef]
- Liparoti, V.; Molinaro, A.; Sturiale, L.; Garozzo, D.; Nazarenko, E.L.; Gorshkova, R.P.; Ivanova, E.P.; Shevcenko, L.S.; Lanzetta, R.; Parrilli, M. Structural analysis of the deep rough lipopolysaccharide from gram negative bacterium alteromonas macleodii atcc 27126t: The first finding of?-kdo in the inner core of lipopolysaccharides. Eur. J. Org. Chem. 2006, 2006, 4710–4716. [Google Scholar] [CrossRef]
- Snyder, D.S.; Brahamsha, B.; Azadi, P.; Palenik, B. Structure of compositionally simple lipopolysaccharide from marine Synechococcus. J. Bacteriol. 2009, 191, 5499–5509. [Google Scholar] [CrossRef]
- Silipo, A.; Leone, S.; Molinaro, A.; Sturiale, L.; Garozzo, D.; Nazarenko, E.L.; Gorshkova, R.P.; Ivanova, E.P.; Lanzetta, R.; Parrilli, M. Complete structural elucidation of a novel lipooligosaccharide from the outer membrane of the marine bacterium Shewanella pacifica. Eur. J. Org. Chem. 2005, 2005, 2281–2291. [Google Scholar]
- Maaetoft-Udsen, K.; Vynne, N.; Heegaard, P.M.; Gram, L.; Frøkiær, H. Pseudoalteromonas strains are potent immunomodulators owing to low-stimulatory LPS. Innate Immun. 2013, 19, 160–173. [Google Scholar] [CrossRef]
- Sinensky, M. Homeoviscous adaptation—A homeostatic process that regulates the viscosity of membrane lipids in Escherichia coli. Proc. Natl. Acad. Sci. USA 1974, 71, 522–525. [Google Scholar] [CrossRef]
- Carty, S.M.; Sreekumar, K.R.; Raetz, C.R.H. Effect of cold shock on lipid a biosynthesis Inescherichia coli: Induction at 12 degrees C of an acyltransferase specific for palmitoleoyl-acyl carrier protein. J. Biol. Chem. 1999, 274, 9677–9685. [Google Scholar]
- Ramos, J.L.; Gallegos, M.T.; Marqués, S.; Ramos-González, M.I.; Espinosa-Urgel, M.; Segura, A. Responses of Gram-negative bacteria to certain environmental stressors. Curr. Opin. Microbiol. 2001, 4, 166–171. [Google Scholar] [CrossRef]
- Peri, F.; Piazza, M. Therapeutic targeting of innate immunity with Toll-like receptor 4 (TLR4) antagonists. Biotechnol. Adv. 2011, 30, 251–260. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef]
- Czeslick, E.; Struppert, A.; Simm, A.; Sablotzki, A. E5564 (eritoran) inhibits lipopolysaccharide-induced cytokine production in human blood monocytes. Inflamm. Res. 2006, 55, 511–515. [Google Scholar] [CrossRef]
- Opal, S.M.; Laterre, P.F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The access randomized trial. JAMA 2013, 309, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Takashima, K.; Matsunaga, N.; Yoshimatsu, M.; Hazeki, K.; Kaisho, T.; Uekata, M.; Hazeki, O.; Akira, S.; Iizawa, Y.; Ii, M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br. J. Pharmacol. 2009, 157, 1250–1262. [Google Scholar] [CrossRef]
- Rooijakkers, S.H.; van Strijp, J.A. Bacterial complement evasion. Mol. Immunol. 2007, 44, 23–32. [Google Scholar] [CrossRef]
- Rooijakkers, S.H.; van Kessel, K.P.; van Strijp, J.A. Staphylococcal innate immune evasion. Trends Microbiol. 2005, 13, 596–601. [Google Scholar] [CrossRef]
- Bardoel, B.W.; van der Ent, S.; Pel, M.J.; Tommassen, J.; Pieterse, C.M.; van Kessel, K.P.; van Strijp, J.A. Pseudomonas evades immune recognition of flagellin in both mammals and plants. PLoS Pathog. 2011, 7, e1002206. [Google Scholar] [CrossRef]
- Laarman, A.J.; Bardoel, B.W.; Ruyken, M.; Fernie, J.; Milder, F.J.; van Strijp, J.A.; Rooijakkers, S.H. Pseudomonas aeruginosa alkaline protease blocks complement activation via the classical and lectin pathways. J. Immunol. (Baltim.) 2012, 188, 386–393. [Google Scholar]
- Bardoel, B.W.; Vos, R.; Bouman, T.; Aerts, P.C.; Bestebroer, J.; Huizinga, E.G.; Brondijk, T.H.; van Strijp, J.A.; de Haas, C.J. Evasion of Toll-like receptor 2 activation by staphylococcal superantigen-like protein 3. J. Mol. Med. (Berl.) 2012, 90, 1109–1120. [Google Scholar] [CrossRef]
- Gomery, K.; Muller-Loennies, S.; Brooks, C.L.; Brade, L.; Kosma, P.; Di Padova, F.; Brade, H.; Evans, S.V. Antibody WN1 222–5 mimics Toll-like receptor 4 binding in the recognition of LPS. Proc. Natl. Acad. Sci. USA 2012, 109, 20877–20882. [Google Scholar] [CrossRef]
- Zhang, S.; Cheng, K.; Wang, X.; Yin, H. Selection, synthesis, and anti-inflammatory evaluation of the arylidene malonate derivatives as tlr4 signaling inhibitors. Bioorg. Med. Chem. 2012, 20, 6073–6079. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Y.; Liu, Y.; Xiang, X.; Dong, Z. Paclitaxel ameliorates lipopolysaccharide-induced kidney injury by binding myeloid differentiation protein-2 to block Toll-like receptor 4-mediated nuclear factor-kappab activation and cytokine production. J. Pharmacol. Exp. Ther. 2013, 345, 69–75. [Google Scholar] [CrossRef]
- Piao, W.; Vogel, S.N.; Toshchakov, V.Y. Inhibition of TLR4 signaling by tram-derived decoy peptides in vitro and in vivo. J. Immunol. (Baltim.) 2013, 190, 2263–2272. [Google Scholar]
- Takahashi, M.; Ota, A.; Karnan, S.; Hossain, E.; Konishi, Y.; Damdindorj, L.; Konishi, H.; Yokochi, T.; Nitta, M.; Hosokawa, Y. Arsenic trioxide prevents nitric oxide production in lipopolysaccharide-stimulated raw 264.7 by inhibiting a TRIF-dependent pathway. Cancer Sci. 2012, 104, 165–170. [Google Scholar]
- Coburn, B.; Sekirov, I.; Finlay, B.B. Type III secretion systems and disease. Clin. Microbiol. Rev. 2007, 20, 535–549. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nijland, R.; Hofland, T.; Van Strijp, J.A.G. Recognition of LPS by TLR4: Potential for Anti-Inflammatory Therapies. Mar. Drugs 2014, 12, 4260-4273. https://doi.org/10.3390/md12074260
Nijland R, Hofland T, Van Strijp JAG. Recognition of LPS by TLR4: Potential for Anti-Inflammatory Therapies. Marine Drugs. 2014; 12(7):4260-4273. https://doi.org/10.3390/md12074260
Chicago/Turabian StyleNijland, Reindert, Tom Hofland, and Jos A. G. Van Strijp. 2014. "Recognition of LPS by TLR4: Potential for Anti-Inflammatory Therapies" Marine Drugs 12, no. 7: 4260-4273. https://doi.org/10.3390/md12074260
APA StyleNijland, R., Hofland, T., & Van Strijp, J. A. G. (2014). Recognition of LPS by TLR4: Potential for Anti-Inflammatory Therapies. Marine Drugs, 12(7), 4260-4273. https://doi.org/10.3390/md12074260