
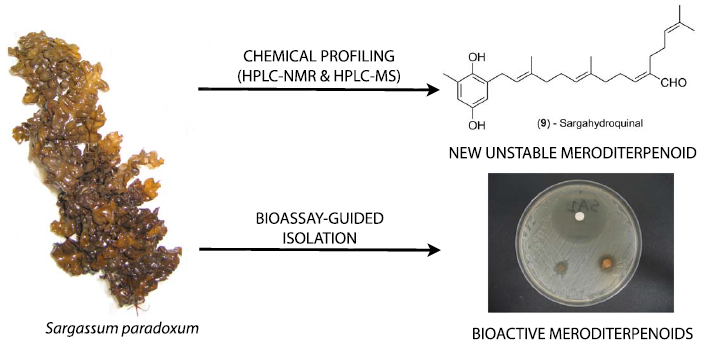
Chemical Profiling (HPLC-NMR & HPLC-MS), Isolation, and Identification of Bioactive Meroditerpenoids from the Southern Australian Marine Brown Alga Sargassum paradoxum


Abstract
:
1. Introduction
2. Results and Discussion

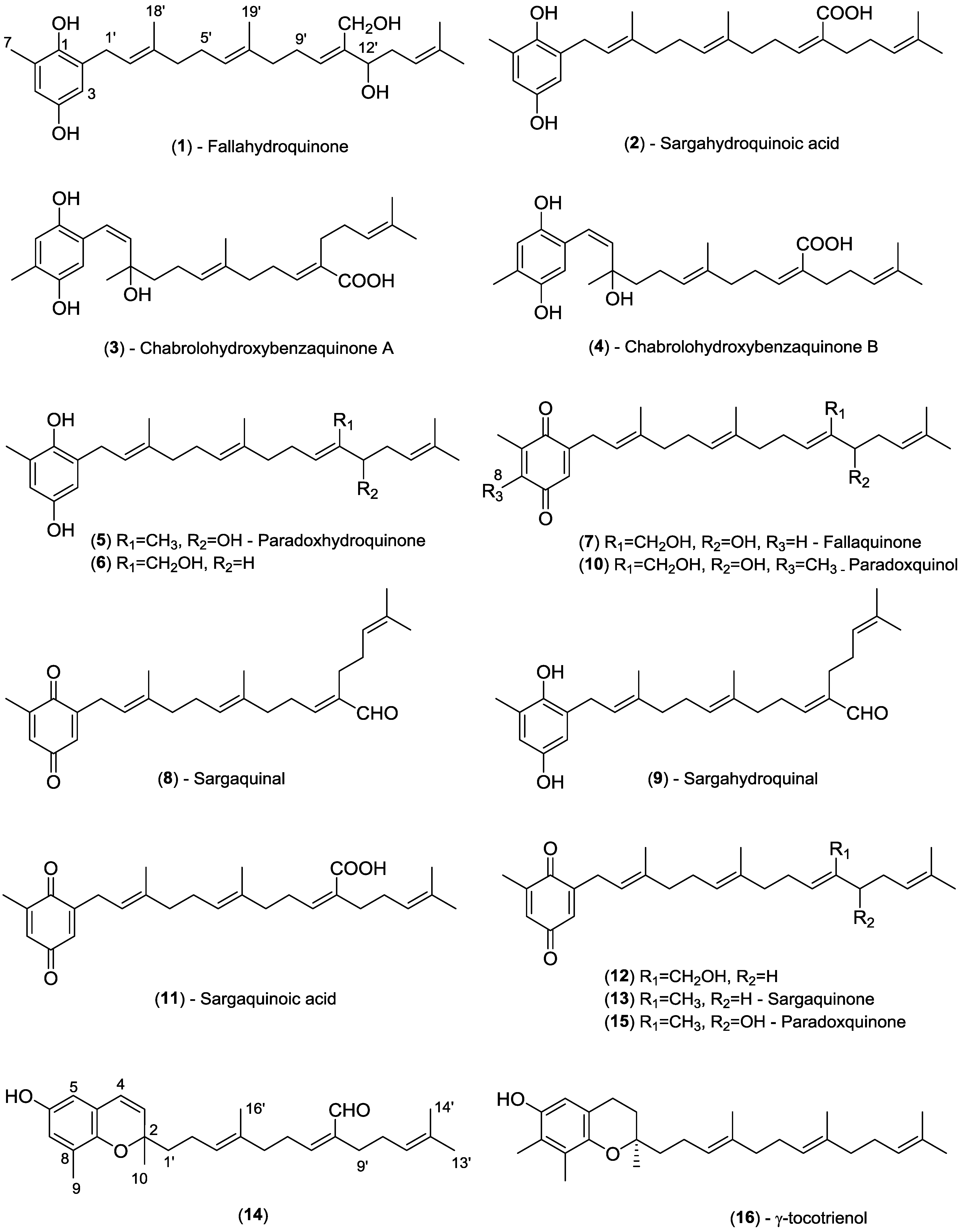
2.1. Chemical Profiling (HPLC-NMR & HPLC-MS)


{kind=link}
{kind=link}
{kind=link}
| Fallahydroquinone (1) | Sargahydroquinoic Acid (2) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | δC a, Type | δH (J in Hz) | gCOSY | gHMBCAD | δC a, Type | δH (J in Hz) | gCOSY | gHMBCAD | nOe | ||||
| 1 | 146.0, C | 146.0, C | |||||||||||
| 2 | ND b | 130.4, C | |||||||||||
| 3 | 114.4, CH | 7.22, d (3.0) | 114.4, CH | 7.23, d (3.0) | 1’ | 1, 4, 5 | |||||||
| 4 | ND b | 150.6, C | |||||||||||
| 5 | 115.6, CH | 7.25, d (3.0) | 7 | 115.6, CH | 7.26, d (3.0) | 7 | 1, 3 | ||||||
| 6 | 126.9, C | 126.9, C | |||||||||||
| 7 | ND b | 2.96, s | 5 | 1, 5, 6 | ND b | 2.96, s | 1, 5, 6 | ||||||
| 1′ | 29.3, CH2 | 4.05, d (7.5) | 2′, 18′ | 1, 3, 2′, 3′ | 29.3, CH2 | 4.06, d (7.0) | 3, 2′, 18′ | 1, 2, 3, 2′, 3′ | |||||
| 2′ | 123.4, CH | 6.10, t (7.5) | 1′, 18′ | 123.4, CH | 6.10, t (7.0) | 1′, 18′ | 1′, 4′, 18′ | ||||||
| 3′ | 137.0, C | 137.1, C | |||||||||||
| 4′ | 40.2, CH2 | SS c | 40.2, CH2 | 2.91, m | |||||||||
| 5′ | ND b | 2.94, m | ND b | 2.94, m | 6′, 19′ | 3′, 4′, 6′, 7′ | |||||||
| 6′ | 125.4, CH | 5.96, t (7.0) | 5′, 19′ | 125.6, CH | 5.97, t (7.0) | 5′, 19′ | 8’, 19’ | ||||||
| 7′ | 135.6, C | 135.4, C | |||||||||||
| 8′ | 40.1, CH2 | SS c | 39.6, CH2 | 2.91, m | 9′ | ||||||||
| 9′ | ND b | 3.01, m | 10′ | 10′, 11′ | 28.4, CH2 | 3.30, dt (7.0, 7.5) | 10′ | 7′, 8′, 10′, 11′ | |||||
| 10′ | 130.7, CH | 6.30, t (7.0) | 9′ | 142.6, CH | 6.69, t (7.5) | 9′ | 8′, 12′, 20′ | 9′, 12′ | |||||
| 11′ | 140.3, C | 132.5, C | |||||||||||
| 12′ | 75.7, CH | 4.87, t (7.5) | 13′ | 10′, 11′ | 35.3, CH2 | 3.02, m | 10′, 11′, 13′, 14′, 20′ | ||||||
| 13′ | 35.4, CH2 | 3.06, dd (7.0,7.5) | 12′, 14′, 16′, 17′ | 12′, 14′, 15′ | 28.3, CH2 | 2.91, m | 17′ | ||||||
| 14′ | 121.6, CH | 5.90, dd (7.0) | 13′ | 124.3, CH | 5.91, t (7.0) | 13′, 16′, 17′ | 16′ | ||||||
| 15′ | 134.1, C | 133.1, C | |||||||||||
| 16′ | 18.0, CH3 | 2.41, s | 14′, 15′, 17′ | 17.7, CH3 | 2.39, s | 14′ | 14′, 15′, 17′ | ||||||
| 17′ | 25.9, CH3 | 2.49, s | 13′ | 14′, 15′, 16′ | 25.7, CH3 | 2.49, s | 14′ | 14′, 15′, 16′ | |||||
| 18′ | 16.2, CH3 | 2.52, s | 1′ | 2′, 3′, 4′ | 16.2, CH3 | 2.53, s | 1′, 2′ | 1′, 2′, 3′, 4′ | |||||
| 19′ | 16.1, CH3 | 2.41, s | 15.9, CH3 | 2.41, s | 6′ | 6′, 7′, 8′ | |||||||
| 20a′ | 57.8, CH2 | 4.95, d (12.5) | 10′ | 170.9, C | |||||||||
| 20b′ | 4.88, d (12.0) | ||||||||||||
| 1-OH | ND b | ND b | |||||||||||
| 4-OH | ND b | ND b | |||||||||||
| 12′-OH | ND b | ||||||||||||
| 20′-OH | ND b | ND b | |||||||||||
| Sargahydroquinal (9) | ||
|---|---|---|
| Position | δH (J in Hz) | gCOSY |
| 1 | ||
| 2 | ||
| 3 | 7.23, d (2.5) | |
| 4 | ||
| 5 | 7.25, d (2.5) | |
| 6 | ||
| 7 | 2.94, s | |
| 1′ | 4.05, d (7.5) | 2′, 18′ |
| 2′ | 6.10, t (7.5) | 1′ |
| 3′ | ||
| 4′ | 2.70-3.00, m | |
| 5′ | 2.96, m | 6′ |
| 6′ | 6.00, t (7.0) | 5′ |
| 7′ | ||
| 8′ | 2.98, m | 9′ |
| 9′ | 3.26, dt (7.0, 8.0) | 8′, 10′ |
| 10′ | 7.37, t (7.0) | 9′ |
| 11′ | ||
| 12′ | 2.70-3.00, m | |
| 13′ | 2.70-3.00, m | |
| 14′ | 5.92, t (7.0) | 16′ |
| 15′ | ||
| 16′ | 2.37, s | 14′ |
| 17′ | 2.48, s | |
| 18′ | 2.52, s | 1′, 2′ |
| 19′ | 2.35, s | |
| 20′ | 10.07, s | |
| 1-OH | ND a | |
| 4-OH | ND a | |
2.2. Off-Line Bioassay-Guided Isolation
| Paradoxhydroquinone (5) | ||||
|---|---|---|---|---|
| Position | δC a, Type | δH (J in Hz) | gCOSY | gHMBCAD |
| 1 | 146.4, C | |||
| 2 | 127.6, C | |||
| 3 | 114.0, CH | 6.46, d (2.5) | 5, 1′ | 1, 4, 5, 1′ |
| 4 | 149.0, C | |||
| 5 | 115.5, CH | 6.50, d (2.5) | 3, 7 | 1, 3, 4, 7 |
| 6 | 125.5, C | |||
| 7 | 16.0, CH3 | 2.18, s | 5 | 1, 5, 6 |
| 8 | ||||
| 1′ | 30.0, CH2 | 3.28, d (7.0) | 3, 2′, 4′w, 18′w | 1, 2, 3, 2′, 3′ |
| 2′ | 122.0, CH | 5.26, t (7.0) | 1′, 4′, 18′ | 1′, 4′, 18′ |
| 3′ | 138.1, C | |||
| 4′ | 39.5, CH2 | 2.08, m | 1′w | 5′, 18′ |
| 5′ | 26.1, CH2 | 2.13, m | 4′, 7′ | |
| 6′ | 124.4, CH | 5.09, m | 4′, 5′, 19′ | |
| 7′ | 135.1, C | |||
| 8′ | 39.2, CH2 | 2.01, m | 6′, 7′, 9′, 19′ | |
| 9′ | 26.1, CH2 | 2.10, m | 8′ | 7′, 10′, 11′ |
| 10′ | 126.2, CH | 5.37, t (6.5) | 9′, 20′ | 12′, 20′ |
| 11′ | 136.6, C | |||
| 12′ | 77.3, CH | 3.97, dd (6.0,7.0) | 13a′, 13b′ | 10′, 14′, 20′ |
| 13a′ | 34.2, CH2 | 2.20, m | 12′, 16′, 17′ | |
| 13b′ | 2.28, ddd (7.0, 8.0, 14.0) | 12′, 16′, 17′ | 12′, 14′, 15′ | |
| 14′ | 120.2, CH | 5.09, m | 16′, 17′ | |
| 15′ | 134.7, C | |||
| 16′ | 18.0, CH3 | 1.63, s | 14′ | 14′, 15′, 17′ |
| 17′ | 25.9, CH3 | 1.72, s | 13b′, 14′ | 14′, 15′, 16′ |
| 18′ | 16.2, CH3 | 1.75, s | 1′, 2′ | 2′, 3′, 4′ |
| 19′ | 16.1, CH3 | 1.59, s | 5′, 6′ | 7′, 8′ |
| 20′ | 11.7, CH3 | 1.61, s | 10′ | 10′, 11′, 12′ |
| 1-OH | ND b | |||
| 4-OH | ND b | |||
| 12′-OH | ND b | |||
| Paradoxquinol (10) | Paradoxquinone (15) | |||||||
|---|---|---|---|---|---|---|---|---|
| Position | δC a, Type | δH (J in Hz) | gCOSY | gHMBCAD | δC a, type | δH (J in Hz) | gCOSY | gHMBCAD |
| 1 | 187.7, C | 188.0, C | ||||||
| 2 | 148.1, C | 148.5, C | ||||||
| 3 | 132.1, CH | 6.46, s | 1′ | 132.3, CH | 6.46, bs | 5, 1′ | 1, 4, 5 | |
| 4 | 187.9, C | 188.0, C | ||||||
| 5 | 140.6, C * | 133.2, CH | 6.54, bs | 3, 7 | 1, 3, 4 | |||
| 6 | 141.5, C * | 145.9, C | ||||||
| 7 | 12.4, CH3 | 2.03, s | 1, 5, 6 | 16.0, CH3 | 2.05, s | 5 | 1, 5, 6 | |
| 8 | 12.1, CH3 | 2.00, s | 4, 5, 6 | 3, 2′, 18′ | 1, 2, 3, 2′, 3′ | |||
| 1′ | 27.4, CH2 | 3.12, d (7.0) | 3, 2′, 18′ | 1, 2, 3, 2′, 3′ | 27.5, CH2 | 3.13, d (7.5) | 1′, 18 | 1′, 4′, 18′ |
| 2′ | 118.2, CH | 5.15, t (7.0) | 1′, 18′ | 4′ | 118.1, CH | 5.15, t (7.5) | ||
| 3′ | 139.5, C | 139.8, C | 2′, 3′, 5′, 18′ | |||||
| 4′ | 39.5, CH2 | 2.06, m | 2′, 5′, 6′, 18′ | 39.6, CH2 | 2.07, m | 6′, 18′w, 19′w | 3′, 4′, 6′, 7′ | |
| 5′ | 26.3, CH2 | 2.12, m | 6′ | 4′, 6′ | 26.2, CH2 | 2.11, m | 5′ | 4′, 5′, 19′ |
| 6′ | 124.4, CH | 5.11, m | 5′, 19′ | 4′, 8′ | 124.2, CH | 5.10, m | ||
| 7′ | 134.7, C | 135.1, C | 6′, 7′, 10′ | |||||
| 8′ | 39.5, CH2 | 2.06, m | 19′ | 39.3, CH2 | 2.02, m | 10′, 20′w | 7′, 10′, 11′ | |
| 9′ | 26.0, CH2 | 2.21, m | 10′ | 26.2, CH2 | 2.11, m | 9′, 20′ | 9′, 12′, 20′ | |
| 10′ | 130.5, CH | 5.53, t (7.0) | 9′ | 12′, 20′ | 126.1, CH | 5.38, t (7.0) | ||
| 11′ | 138.9, C | 136.7, C | 13a′ | 10′, 11′, 13′, 14′, 20′ | ||||
| 12′ | 76.9, CH | 4.16, dd (5.5, 7.5) | 13a′, 13b′ | 10′ | 77.2, CH | 3.97, dd (5.5, 7.5) | 12′, 17′ | |
| 13a′ | 35.1, CH2 | 2.26, m | 12′, 14′ | 34.2, CH2 | 2.20, m | 12′, 17′ | 11′, 12′, 14′, 15′ | |
| 13b′ | 2.43, ddd (7.5, 8.5, 14.5) | 12′, 13a′, 14′ | 12′, 14′ | 2.27, m | 17′ | |||
| 14′ | 119.9, CH | 5.11, m | 16′, 17′ | 120.3, CH | 5.09, m | |||
| 15′ | 135.4, C | 134.6, C | 14′, 15′ | |||||
| 16′ | 18.1, CH3 | 1.65, s | 13a′, 13b′ | 14′, 15′, 17′ | 18.0, CH3 | 1.63, s | 14′, 15′, 16′ | |
| 17′ | 25.9, CH3 | 1.73, s | 13a′, 13b′, 14′ | 14′, 15′, 16′ | 25.9, CH3 | 1.72, s | 2′, 3′ | |
| 18′ | 16.1, CH3 | 1.62, s | 1′, 2′ | 2′, 3′, 4′ | 16.1, CH3 | 1.61, s | 7′ | |
| 19′ | 16.1, CH3 | 1.60, s | 5′, 6′ | 6′, 7′, 8′ | 16.1, CH3 | 1.61, s | 10′, 11′ | |
| 20′ | 58.5, CH2 | 4.26, d (3.5) | 10′, 11′. 12′ | 11.7, CH3 | 1.61, s | |||
| 12′-OH | ND b | ND b | ||||||
| 20′-OH | ND b | |||||||
2.3. Antimicrobial Studies
3. Experimental Section
3.1. General Experimental Procedures
3.2. Biological Evaluation
3.3. Marine Alga Material
3.4. Chemical Profiling
| Microorganism | E. coli | S. aureus | S. aureus MRSA | P. aeruginosa | S. pyogenes | C. albicans | |
|---|---|---|---|---|---|---|---|
| Concentration (mg/mL) | ATCC 25922 | ATCC 25923 | 344/2-32 | ATCC 27853 | 345/1 | ATCC 10231 | |
| Dichloromethane extract | 50 | ND g | 2 | 1 | ND g | 5 | ND g |
| Methanol extract | 50 | ND g | 1 | 1 | 10 | 3 | 4 |
| Silica fraction 1 a | 25 | ND g | ND g | ND g | ND g | ND g | ND g |
| Silica fraction 4 b | 25 | ND g | ND g | ND g | ND g | 1 | ND g |
| Silica fraction 6 c | 50 | ND g | ND g | 1 | ND g | 1 | ND g |
| Silica fraction 10 d | 25 | ND g | ND g | 1 | 4 | 3 | ND g |
| Silica fraction 13 e | 50 | ND g | ND g | 1 | ND g | 3 | ND g |
| Silica fraction 17 f | 50 | ND g | ND g | ND g | ND g | ND g | ND g |
| Fallahydroquinone (1) | 1 | ND g | ND g | ND g | ND g | 1 | ND g |
| Sargahydroquinoic acid (2) | 1 | ND g | 1 | 1 | 2 | 3 | ND g |
| Paradoxhydroquinone (5) & compound (6) | 1 | ND g | ND g | ND g | ND g | 3 | ND g |
| Fallaquinone (7) | 1 | ND g | ND g | ND g | 4 | ND g | ND g |
| Sargaquinal (8) | 1 | ND g | ND g | ND g | 3 | 1 | ND g |
| Paradoxquinol (10) | 1 | ND g | ND g | ND g | ND g | 1 | ND g |
| Sargaquinoic acid (11) | 1 | ND g | 1 | 1 | 1 | 3 | ND g |
| Paradoxquinone (15) & compound (12) | 1 | ND g | ND g | ND g | ND g | 2 | ND g |
| Sargaquinone (13) | 1 | ND g | ND g | ND g | 5 | ND g | ND g |
| Ampicillin (antibiotic) | 1 | ND g | 15 | 3 | 2 | 20 | NT h |
| Carbendazim (antifungal) | 1 | NT h | NT h | NT h | NT h | NT h | ND g |
3.5. Extraction and Isolation
3.6. On-Line (HPLC-NMR & HPLC-MS) Characterization of Compounds
3.7. Off-Line Characterization of Compounds
4. Conclusions
Acknowledgments
Author Contributions
Supplementary Information
Conflicts of Interest
References
- MarinLit Database; Royal Society of Chemistry: Cambridge, UK, 2014.
- Guiry, M.D.; Guiry, G.M. AlgaeBase; World-Wide Electronic Publication: National University of Ireland, Galway, Ireland, 2014; Available online: http://www.algaebase.org (accessed on 1 December 2014).
- Phillips, N. Biogeography of Sargassum (Phaeophyta) in the Pacfic basin. Taxon. Ecomonic Seaweeds 1995, 5, 107–144. [Google Scholar]
- Dixon, R.R.M.; Mattio, L.; Huisman, J.M.; Payri, C.E.; Bolton, J.J.; Gurgel, C.F.D. North meets south—Taxonomic and biogeographic implications of a phylogenetic assessment of Sargassum subgenera Arthrophycus and Bactrophycus (Fucales, Phaeophyceae). Phycologia 2014, 53, 15–22. [Google Scholar] [CrossRef]
- Liu, L.; Heinrich, M.; Myers, S.; Dworjanyn, S.A. Towards a better understanding of medicinal uses of the brown seaweed Sargassum in traditional chinese medicine: A phytochemical and pharmacological review. J. Ethnopharmacol. 2012, 142, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Jang, K.H.; Kim, B.; Lee, B.H.; Choi, B.W.; Oh, K.-B.; Shin, J. Meroditerpenoids from the brown alga Sargassum siliquastrum. J. Nat. Prod. 2008, 71, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Urban, S. Meroditerpenoids from the southern australian marine brown alga Sargassum fallax. Phytochemistry 2009, 70, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Segawa, M.; Shirahama, H. New plastoquinones from the brown alga Sargassum sagamianum var. yezoense. Chem. Lett. 1987, 16, 1365–1366. [Google Scholar] [CrossRef]
- Kim, M.C.; Kwon, H.C.; Kim, S.N.; Kim, H.S.; Um, B.H. Plastoquinones from Sargassum yezoense; chemical structures and effects of the activation of peroxisome proliferator-activated receptor gamma. Chem. Pharm. Bull. 2011, 59, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; Tsutsumi, S.; Takada, Y.; Kimura, J. Antibacterial quinone metabolites from the brown alga, Sargassum sagamianum. Bull. Chem. Soc. Jpn. 2008, 81, 1125–1130. [Google Scholar] [CrossRef]
- Culioli, G.; Ortalo-Magne, A.; Valls, R.; Hellio, C.; Clare, A.S.; Piovetti, L. Antifouling activity of meroditerpenoids from the marine brown alga Halidrys siliquosa. J. Nat. Prod. 2008, 71, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Foti, M.; Piattelli, M.; Amico, V.; Ruberto, G. Antioxidant activity of phenolic meroditerpenoids from marine algae. J. Photochem. Photobiol. B 1994, 26, 159–164. [Google Scholar] [CrossRef]
- SciFinder; American Chemical Society: Washington, DC, USA, 2014.
- Scott, A.I. Interpretation of the Ultraviolet Spectra of Natural Products; Pergamon Press Ltd.: London, UK, 1964. [Google Scholar]
- Sheu, J.-H.; Su, J.-H.; Sung, P.-J.; Wang, G.-H.; Dai, C.-F. Novel meroditerpenoid-related metabolites from the formosan soft coral Nephthea chabrolii. J. Nat. Prod. 2004, 67, 2048–2052. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Shindo, T.; Hasegawa, K.; Ushiyama, S. Preparation of 2-(11-Hydroxymethyl-3,7,15-Trimethyl-2,6,10,14-Hexadecatetraenyl)-6-Methyl-1,4-Benzoquinone or -1,4-Benzenediol or Their Acyl Derivatives as 5-Lipoxygenase Inhibitors. Kokai Tokkyo Koho Japan H02-290826, 1990. [Google Scholar]
- Kusumi, T.; Shibata, Y.; Ishitsuka, M.; Kinoshita, T.; Kakisawa, H. Structures of new plastoquinones from the brown alga Sargassum serratifolium. Chem. Lett. 1979, 8, 277–278. [Google Scholar] [CrossRef]
- Amico, V.; Cunsolo, F.; Piattelli, M.; Ruberto, G. Tetraprenyltoluquinols from the brown alga Cystoseira jabukae. Phytochemistry 1985, 24, 1047–1050. [Google Scholar] [CrossRef]
- Kimura, J.; Horie, C.; Marushima, H.; Matsumoto, Y.; Sanjoba, C.; Osada, Y. Antileishmanial Agents Containing p-Benzoquinone Derivative. JP 2012-502793, 2012. [Google Scholar]
- Silva, D.H.S.; Pereira, F.C.; Zanoni, M.V.B.; Yoshida, M. Lipophyllic antioxidants from Iryanthera juruensis fruits. Phytochemistry 2001, 57, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Barr, R.; Crane, F.L. Comparative studies on plastoquinones. III. Distribution of plastoquinones in higher plants. Plant Physiol. 1967, 42, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Gonnella, N.C.; Nakanishi, K. General method for determining absolute configurations of acyclic allylic alcohols. J. Am. Chem. Soc. 1982, 104, 3775–3776. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ito, M.; Koyama, T.; Ogura, K. Stereochemistry of the carbon to carbon bond formation in the biosynthesis of polyprenyl chains with Z double bonds. Studies with undecaprenyl-pyrophosphate synthetase. J. Am. Chem. Soc. 1985, 107, 4588–4589. [Google Scholar] [CrossRef]
- Lamshoft, M.; Schmickler, H.; Marner, F.-J. Determination of the absolute configuration of hydroxyiridals by chiroptical and NMR spectroscopic methods. Eur. J. Org. Chem. 2003, 2003, 727–733. [Google Scholar] [CrossRef]
- Sunazuka, T.; Shirahata, T.; Yoshida, K.; Yamamoto, D.; Harigaya, Y.; Nagai, T.; Kiyohara, H.; Yamada, H.; Kuwajima, I.; Omura, S. Total synthesis of pinellic acid, a potent oral adjuvant for nasal influenza vaccine. Determination of the relative and absolute configuration. Tetrahedron Lett. 2002, 43, 1265–1268. [Google Scholar] [CrossRef]
- De la Mare, J.-A.; Lawson, J.C.; Chiwakata, M.T.; Beukes, D.R.; Edkins, A.L.; Blatch, G.L. Quinones and halogenated monoterpenes of algal origin show anti-proliferative effects against breast cancer cells in vitro. Investig. New Drugs 2012, 30, 2187–2200. [Google Scholar]
- Seo, Y.; Lee, H.-J.; Park, K.E.; Kim, Y.A.; Ahn, J.W.; Yoo, J.S.; Lee, B.-J. Peroxynitrite-scavenging constituents from the brown alga Sargassum thunbergii. Biotechnol. Bioprocess Eng. 2004, 9, 212–216. [Google Scholar] [CrossRef]
- Afolayan, A.F.; Bolton, J.J.; Lategan, C.A.; Smith, P.J.; Beukes, D.R. Fucoxanthin, tetraprenylated toluquinone, and toluhydroquinone metabolites from Sargassum heterophyllum inhibit the in vitro growth of the malaria parasite Plasmodium falciparum. J. Biosci. 2008, 63, 848–852. [Google Scholar]
- Cespedes, C.L.; Torres, P.; Marin, J.C.; Arciniegas, A.; de Vivar, A.R.; Perez-Castorena, A.L.; Aranda, E. Insect growth inhibition by tocotrienols and hydroquinones from Roldana barba-johannis. Phytochemistry 2004, 65, 1963–1975. [Google Scholar] [CrossRef] [PubMed]
- Park, B.-G.; Shin, W.-S.; Um, Y.; Cho, U.; Park, G.-M.; Yeon, D.-S.; Kwon, S.-C.; Ham, J.; Choi, B.W.; Lee, S. Selective vasodilatation effect of sargahydroquinoic acid, an active constituent of Sargassum micracanthum, on the basilar arteries of rabbits. Bioorg. Med. Chem. Lett. 2008, 18, 2624–2627. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-N.; Choi, H.Y.; Lee, W.; Park, G.-M.; Shin, W.-S.; Kim, Y.K. Sargaquinoic acid and sargahydroquinoic acid from Sargassum yezoense stimulate adipocyte differentiation through PPARα/γ activation in 3T3-L1 cells. FEBS Lett. 2008, 582, 3465–3472. [Google Scholar]
- Kamei, Y.; Tsang, C.K. Sargaquinoic acid promotes neurite outgrowth via protein kinase A and MAP kinases-mediated signaling pathways in PC12D cells. Int. J. Dev. Neurosci. 2003, 21, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Hur, S.; Lee, H.; Kim, Y.; Lee, B.-H.; Shin, J.; Kim, T.-Y. Sargaquinoic acid and sargachromenol, extracts of Sargassum sagamianum, induce apoptosis in HaCaT cells and mice skin: Its potentiation of UVB-induced apoptosis. Eur. J. Pharmacol. 2008, 582, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.H.S.; Zhang, Y.; Santos, L.A.; Bolzani, V.S.; Nair, M.G. Lipoperoxidation and cyclooxygenases 1 and 2 inhibitory compounds from Iryanthera juruensis. J. Agric. Food Chem. 2007, 55, 2569–2574. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.W.; Ryu, G.; Park, S.H.; Kim, E.S.; Shin, J.; Roh, S.S.; Shin, H.C.; Lee, B.-H. Anticholinesterase activity of plastoquinones from Sargassum sagamianum: Lead compounds for Alzheimer’s disease therapy. Phytother. Res. 2007, 21, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Nahas, R.; Abatis, D.; Anagnostopoulou, M.A.; Kefalas, P.; Vagias, C.; Roussis, V. Radical-scavenging activity of aegean sea marine algae. Food Chem. 2007, 102, 577–581. [Google Scholar] [CrossRef]
- Tziveleka, L.-A.; Abatis, D.; Paulus, K.; Bauer, R.; Vagias, C.; Roussis, V. Marine polyprenylated hydroquinones, quinones and chromenols with inhibitory effects on leukotriene formation. Chem. Biodivers. 2005, 2, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Voutquenne, L.; Lavaud, C.; Massiot, G.; Sevenet, T.; Hadi, H.A. Cytotoxic polyisoprenes and glycosides of long-chain fatty alcohols from Dimocarpus fumatus. Phytochemistry 1999, 50, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Fenical, W.; Norris, J.N. Chemical variation in the tropical seaweed Stypopodium zonale (dictyotaceae). Phytochemistry 1985, 24, 1279–1283. [Google Scholar] [CrossRef]
- Master, R.N.; Clark, R.B.; Karlowsky, J.A.; Ramirez, J.; Bordon, J.M. Analysis of resistance, cross-resistance and antimicrobial combinations for Pseudomonas aeruginosa isolates from 1997 to 2009. Int. J. Antimicrob. Agents 2011, 38, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Smallcombe, S.H.; Patt, S.L.; Keifer, P.A. WET solvent suppression and its applications to LC NMR and high-resolution NMR spectroscopy. J. Magn. Reson. Ser. A 1995, 117, 295–303. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brkljača, R.; Urban, S. Chemical Profiling (HPLC-NMR & HPLC-MS), Isolation, and Identification of Bioactive Meroditerpenoids from the Southern Australian Marine Brown Alga Sargassum paradoxum. Mar. Drugs 2015, 13, 102-127. https://doi.org/10.3390/md13010102
Brkljača R, Urban S. Chemical Profiling (HPLC-NMR & HPLC-MS), Isolation, and Identification of Bioactive Meroditerpenoids from the Southern Australian Marine Brown Alga Sargassum paradoxum. Marine Drugs. 2015; 13(1):102-127. https://doi.org/10.3390/md13010102
Chicago/Turabian StyleBrkljača, Robert, and Sylvia Urban. 2015. "Chemical Profiling (HPLC-NMR & HPLC-MS), Isolation, and Identification of Bioactive Meroditerpenoids from the Southern Australian Marine Brown Alga Sargassum paradoxum" Marine Drugs 13, no. 1: 102-127. https://doi.org/10.3390/md13010102
APA StyleBrkljača, R., & Urban, S. (2015). Chemical Profiling (HPLC-NMR & HPLC-MS), Isolation, and Identification of Bioactive Meroditerpenoids from the Southern Australian Marine Brown Alga Sargassum paradoxum. Marine Drugs, 13(1), 102-127. https://doi.org/10.3390/md13010102