
Marine Compound Xyloketal B Reduces Neonatal Hypoxic-Ischemic Brain Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results and Discussion
2.1. Xyloketal B Reduces OGD-Induced Cell Death in Primary Cortical Culture

2.2. Xyloketal B Reduces Calcium Entry in Primary Cortical Neurons
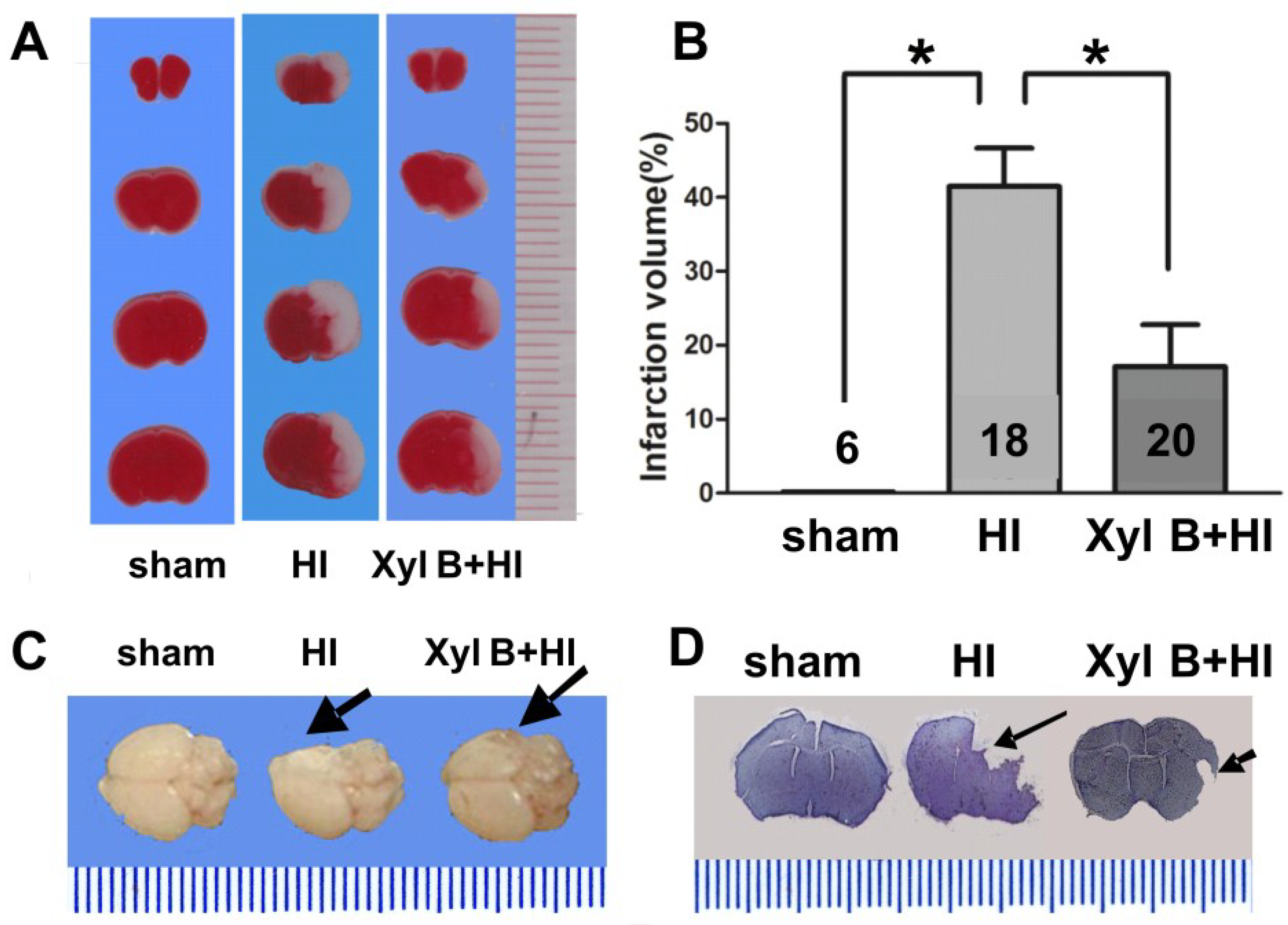
2.3. Xyloketal B Reduces Hypoxic-Ischemic Brain Injury and Improved Behavioral Performance

2.3.1. Infarct Volume

2.3.2. Body Weight

2.3.3. Neurobehavioral Tests
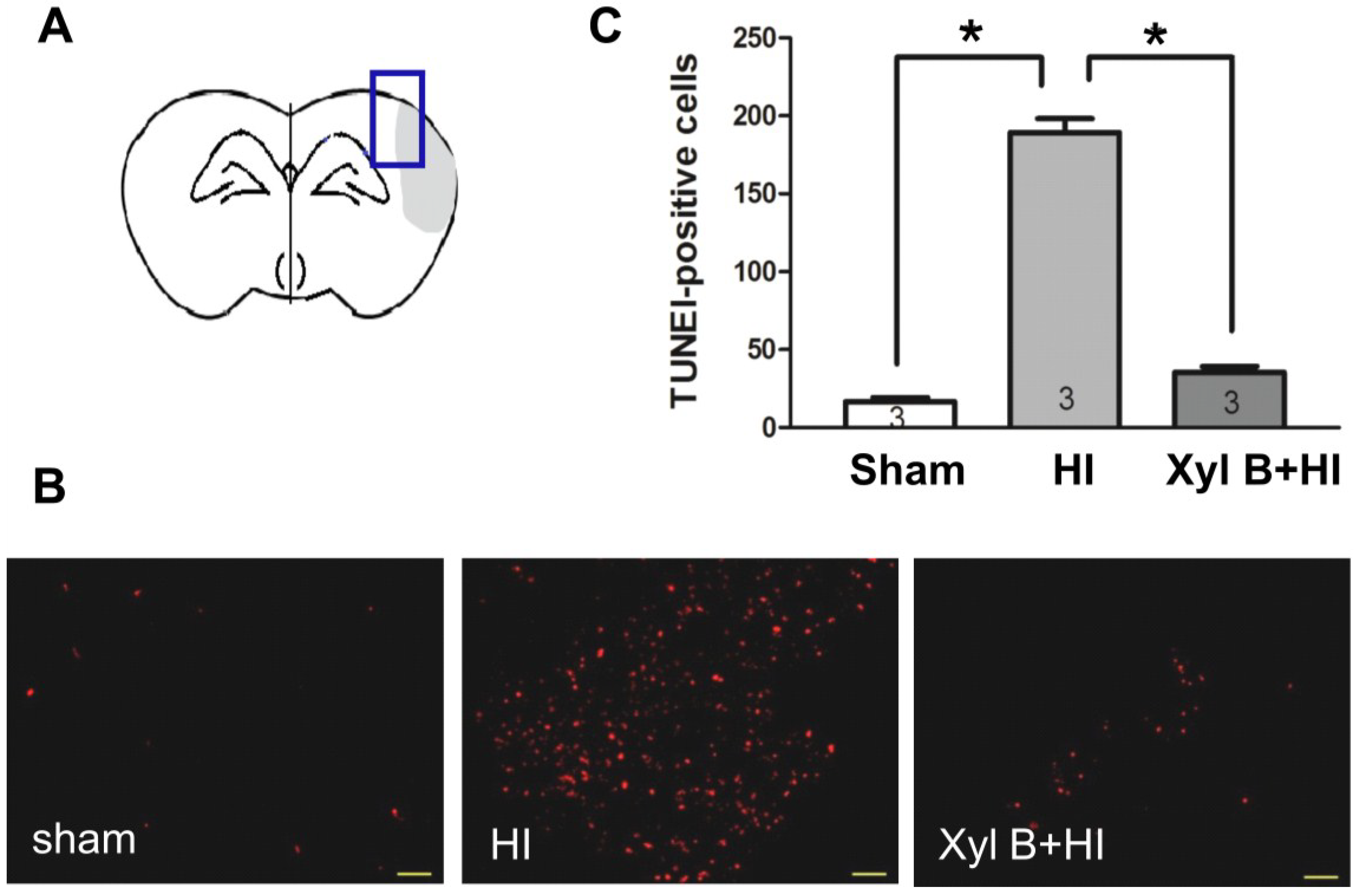
2.4. Effects of Xyloketal B on Apoptosis
2.4.1. Xyloketal B Inhibits HI-induced Apoptosis

2.4.2. Xyloketal B Inhibits the Expression of Apoptosis-Associated Proteins

2.5. Discussion
3. Experimental Section
3.1. Animals
3.2. Reagents and Drugs
3.3. Primary Cell Culture and Oxygen-Glucose Deprivation (OGD) of Cultured Cortical Neurons
3.4. Fura-2 Calcium Imaging
3.5. Drug Administration
3.6. Hypoxic-Ischemic Brain Injury Model
3.7. Functional Recovery Assessments
3.8. General Histology and Infarct Size Measurement
3.9. Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling (TUNEL) Assay
3.10. Western Blots Analysis
3.11. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| DAPI | 4′,6-diamidino-2-phenylindole |
| DIV | day in vitro |
| DMSO | dimethyl sulfoxide |
| MPP+ | 1-methyl-4-phenylpyridinium |
| OGD | oxygen-glucose deprivation |
| PI | propidium iodide |
| PVDF | polyvinylidene difluoride |
Conflicts of Interest
References
- Nelson, K.B. Neonatal encephalopathy: Etiology and outcome. Dev. Med. Child Neurol. 2005, 47, 292. [Google Scholar] [CrossRef] [PubMed]
- Schendel, D.; Nelson, K.B.; Blair, E. Neonatal encephalopathy or hypoxic-ischemic encephalopathy? Ann. Neurol. 2012, 72, 984–985. [Google Scholar] [CrossRef] [PubMed]
- Lawn, J.E.; Cousens, S.; Zupan, J. 4 million neonatal deaths: When? Where? Why? Lancet 2005, 365, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Pierrat, V.; Haouari, N.; Liska, A.; Thomas, D.; Subtil, D.; Truffert, P. Prevalence, causes, and outcome at 2 years of age of newborn encephalopathy: Population based study. Arch. Dis. Child. Fetal Neonatal Ed. 2005, 90, F257–F261. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, J.D.; Wilson, P.D. Synthesis of xyloketal A, B, C, D, and G analogues. J. Org. Chem. 2006, 71, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, Y.; Xiang, Q.; Pei, Z.; Liu, X.; Lu, B.; Chen, L.; Wang, G.; Pang, J.; Lin, Y. Design and synthesis of novel xyloketal derivatives and their vasorelaxing activities in rat thoracic aorta and angiogenic activities in zebrafish angiogenesis screen. J. Med. Chem. 2010, 53, 4642–4653. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, Y.; Wang, J.; Pang, J.; Huang, C.; Wu, X.; She, Z.; Vrijmoed, L.L.; Jones, E.B.; Lin, Y. Benzofuran derivatives from the mangrove endophytic Fungus Xylaria sp. (#2508). J. Nat. Prod. 2008, 71, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, X.; Feng, S.; Jiang, G.; Luo, J.; Zhou, S.; Vrijmoed, L.L.; Jones, E.B.; Krohn, K.; Steingrover, K.; et al. Five unique compounds: Xyloketals from mangrove fungus Xylaria sp. from the South China Sea coast. J. Org. Chem. 2001, 66, 6252–6256. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Qian, Y.; Meng, W.F.; Pang, J.Y.; Lin, Y.C.; Guan, Y.Y.; Chen, S.P.; Liu, J.; Pei, Z.; Wang, G.L. A novel marine compound xyloketal B protects against oxidized LDL-induced cell injury in vitro. Biochem. Pharmacol. 2009, 78, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Chen, J.W.; Yuan, F.; Huang, Y.Y.; Zhao, L.Y.; Li, J.; Su, H.X.; Liu, J.; Pang, J.Y.; Lin, Y.C.; et al. Xyloketal B exhibits its antioxidant activity through induction of HO-1 in vascular endothelial cells and zebrafish. Mar. Drugs 2013, 11, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.L.; Yao, X.L.; Liu, Z.; Zhang, H.; Li, W.; Li, Z.; Wang, G.L.; Pang, J.; Lin, Y.; Xu, Z.; et al. Protective effects of xyloketal B against MPP+-induced neurotoxicity in Caenorhabditis elegans and PC12 cells. Brain Res. 2010, 1332, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, C.; Guo, W.; Zhang, X.; Liu, S.; Liang, F.; Xu, Z.; Pei, Z.; Song, H.; Qiu, L.; et al. Synthesis and neuroprotective action of xyloketal derivatives in Parkinson’s disease models. Mar. Drugs 2013, 11, 5159–5189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, L.; Ling, C.; Li, J.; Pang, J.Y.; Lin, Y.C.; Liu, J.; Huang, R.; Wang, G.L.; Pei, Z.; et al. Marine compound Xyloketal B protects PC12 cells against OGD-induced cell damage. Brain Res. 2009, 1302, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.; Soriano, M.; Blomgren, K.; Hagberg, H.; Wybrecht, R.; Miss, M.T.; Hoefer, S.; Adam, G.; Niederhauser, O.; Kemp, J.A.; et al. Role of caspase-3 activation in cerebral ischemia-induced neurodegeneration in adult and neonatal brain. J. Cereb. Blood Flow Metable 2002, 22, 420–430. [Google Scholar] [CrossRef]
- Wang, X.; Carlsson, Y.; Basso, E.; Zhu, C.; Rousset, C.I.; Rasola, A.; Johansson, B.R.; Blomgren, K.; Mallard, C.; Bernardi, P.; et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J. Neurosci. 2009, 29, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Bdel-Hamid, K.M.; Tymianski, M. Mechanisms and effects of intracellular calcium buffering on neuronal survival in organotypic hippocampal cultures exposed to anoxia/aglycemia or to excitotoxins. J. Neurosci. 1997, 17, 3538–3553. [Google Scholar] [PubMed]
- Gendron, T.F.; Mealing, G.A.; Paris, J.; Lou, A.; Edwards, A.; Hou, S.T.; MacManus, J.P.; Hakim, A.M.; Morley, P. Attenuation of neurotoxicity in cortical cultures and hippocampal slices from E2F1 knockout mice. J. Neurochem. 2001, 78, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Xiao, M.; Sun, F.; Xiao, X.; Pei, W.; Li, J.; Graham, S.H.; Simon, R.P.; Chen, J. Cloning of a novel Apaf-1-interacting protein: A potent suppressor of apoptosis and ischemic neuronal cell death. J. Neurosci. 2004, 24, 6189–6201. [Google Scholar] [CrossRef] [PubMed]
- Alibrahim, A.; Zhao, L.Y.; Bae, C.Y.; Barszczyk, A.; Sun, C.L.; Wang, G.L.; Sun, H.S. Neuroprotective effects of volume-regulated anion channel blocker DCPIB on neonatal hypoxic-ischemic injury. Acta Pharmacol. Sin. 2013, 34, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Levine, S. Anoxic-ischemic encephalopathy in rats. Am. J. Pathol. 1960, 36, 1–17. [Google Scholar] [PubMed]
- Rice, J.E., III; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.Y.; Ashwal, S. Animal models of perinatal hypoxic-ischemic brain damage. Pediatr. Neurol. 2009, 40, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, R.C.; Vannucci, S.J. Perinatal hypoxic-ischemic brain damage: Evolution of an animal model. Dev. Neurosci. 2005, 27, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Zhai, L.; Feng, X.; Chen, J.; Miao, Z.; Ren, L.; Qian, X.; Yu, J.; Li, Y.; Xu, X.; et al. Apelin-36, a potent peptide, protects against ischemic brain injury by activating the PI3K/Akt pathway. Neurochem. Int. 2013, 63, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Jantas, D.; Lason, W. Different mechanisms of NMDA-mediated protection against neuronal apoptosis: A stimuli-dependent effect. Neurochem. Res. 2009, 34, 2040–2054. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Mallard, C.; Rousset, C.I.; Xiaoyang, W. Apoptotic mechanisms in the immature brain: Involvement of mitochondria. J. Child Neurol. 2009, 24, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Xu, F.; Bahr, B.A.; Shibata, M.; Uchiyama, Y.; Hagberg, H.; Blomgren, K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005, 12, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Sastry, P.S.; Rao, K.S. Apoptosis and the nervous system. J. Neurochem. 2000, 74, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Northington, F.J.; Graham, E.M.; Martin, L.J. Apoptosis in perinatal hypoxic-ischemic brain injury: How important is it and should it be inhibited? Brain Res. Rev. 2005, 50, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Baydas, G.; Reiter, R.J.; Akbulut, M.; Tuzcu, M.; Tamer, S. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and by regulating pro- and anti-apoptotic protein levels. Neuroscience 2005, 135, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Lu, M.; Li, Q.J.; Zhang, Z.; Wu, Z.Z.; Li, J.; Qian, L.; Xu, Y.; Wang, Z.Y. Hyperbaric Oxygen Suppresses Hypoxic-Ischemic Brain Damage in Newborn Rats. J. Child Neurol. 2014, in press. [Google Scholar]
- Muller, M.M.; Middelanis, J.; Meier, C.; Surbek, D.; Berger, R. 17beta-estradiol protects 7-day old rats from acute brain injury and reduces the number of apoptotic cells. Reprod. Sci. 2013, 20, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Chauvier, D.; Renolleau, S.; Holifanjaniaina, S.; Ankri, S.; Bezault, M.; Schwendimann, L.; Rousset, C.; Casimir, R.; Hoebeke, J.; Smirnova, M.; et al. Targeting neonatal ischemic brain injury with a pentapeptide-based irreversible caspase inhibitor. Cell Death Dis. 2011, 2, e203. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Sun, Y.; Wang, X.; Zhu, C.; Blomgren, K. Delayed, long-term administration of the caspase inhibitor Q-VD-OPh reduced brain injury induced by neonatal hypoxia-ischemia. Dev. Neurosci. 2014, 36, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E. Neural apoptosis. Ann. Neurol. 1995, 38, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Won, C.K.; Kim, M.O.; Koh, P.O. Estrogen modulates Bcl-2 family proteins in ischemic brain injury. J. Vet. Med. Sci. 2006, 68, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Fujimura, M.; Morita-Fujimura, Y.; Kawase, M.; Chan, P.H. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J. Neurosci. 1999, 19, RC39. [Google Scholar] [PubMed]
- Ouyang, Y.B.; Tan, Y.; Comb, M.; Liu, C.L.; Martone, M.E.; Siesjo, B.K.; Hu, B.R. Survival- and death-promoting events after transient cerebral ischemia: Phosphorylation of Akt, release of cytochrome C and Activation of caspase-like proteases. J. Cereb. Blood Flow Metable 1999, 19, 1126–1135. [Google Scholar] [CrossRef]
- Hetz, C.; Vitte, P.A.; Bombrun, A.; Rostovtseva, T.K.; Montessuit, S.; Hiver, A.; Schwarz, M.K.; Church, D.J.; Korsmeyer, S.J.; Martinou, J.C.; et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J. Biol. Chem. 2005, 280, 42960–42970. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.; Zhang, Y.; Xiang, J.; Zhang, W.; Wang, G.H.; Li, W.W.; Xu, L.L.; Cai, D.F. Xiao-Xu-Ming decoction preserves mitochondrial integrity and reduces apoptosis after focal cerebral ischemia and reperfusion via the mitochondrial p53 pathway. J. Ethnopharmacol. 2014, 151, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Yuan, Z.Y.; Wang, Y.G.; Wan, H.J.; Hu, J.; Chai, Y.S.; Lei, F.; Xing, D.M.; DU, L.J. Role of baicalin in regulating Toll-like receptor 2/4 after ischemic neuronal injury. Chin. Med. J. (Engl.) 2012, 125, 1586–1593. [Google Scholar]
- Green, D.R. Apoptotic pathways: Paper wraps stone blunts scissors. Cell 2000, 102, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Iihara, K.; Wei, W.L.; Xiong, Z.G.; Arundine, M.; Cerwinski, W.; MacDonald, J.F.; Tymianski, M. A key role for TRPM7 channels in anoxic neuronal death. Cell 2003, 115, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef] [PubMed]
- Sanches, E.F.; Arteni, N.S.; Spindler, C.; Moyses, F.; Siqueira, I.R.; Perry, M.L.; Netto, C.A. Effects of pre- and postnatal protein malnutrition in hypoxic-ischemic rats. Brain Res. 2012, 1438, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Bouslama, M.; Renaud, J.; Olivier, P.; Fontaine, R.H.; Matrot, B.; Gressens, P.; Gallego, J. Melatonin prevents learning disorders in brain-lesioned newborn mice. Neuroscience 2007, 150, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Yan, H.; Xu, M.; Zhao, Y.L.; Li, L.M.; Zhou, X.H.; Wang, M.X.; Ma, L. Hyperbaric oxygenation reduces long-term brain injury and ameliorates behavioral function by suppression of apoptosis in a rat model of neonatal hypoxia-ischemia. Neurochem. Int. 2013, 62, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Nejatbakhsh, N.; Guo, C.H.; Lu, T.Z.; Pei, L.; Smit, A.B.; Sun, H.S.; van Kesteren, R.E.; Feng, Z.P. Caltubin, a novel molluscan tubulin-interacting protein, promotes axonal growth and attenuates axonal degeneration of rodent neurons. J. Neurosci. 2011, 31, 15231–15244. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.S.; Jackson, M.F.; Martin, L.J.; Jansen, K.; Teves, L.; Cui, H.; Kiyonaka, S.; Mori, Y.; Jones, M.; Forder, J.P.; et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat. Neurosci. 2009, 12, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Gardzinski, P.; Lee, D.W.; Fei, G.H.; Hui, K.; Huang, G.J.; Sun, H.S.; Feng, Z.P. The role of synaptotagmin I C2A calcium-binding domain in synaptic vesicle clustering during synapse formation. J. Physiol. 2007, 581, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.; Fei, G.H.; Saab, B.J.; Su, J.; Roder, J.C.; Feng, Z.P. Neuronal calcium sensor-1 modulation of optimal calcium level for neurite outgrowth. Development 2007, 134, 4479–4489. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, A.-J.; Chen, W.; Xu, B.; Liu, R.; Turlova, E.; Barszczyk, A.; Sun, C.L.; Liu, L.; Deurloo, M.; Wang, G.-L.; et al. Marine Compound Xyloketal B Reduces Neonatal Hypoxic-Ischemic Brain Injury. Mar. Drugs 2015, 13, 29-47. https://doi.org/10.3390/md13010029
Xiao A-J, Chen W, Xu B, Liu R, Turlova E, Barszczyk A, Sun CL, Liu L, Deurloo M, Wang G-L, et al. Marine Compound Xyloketal B Reduces Neonatal Hypoxic-Ischemic Brain Injury. Marine Drugs. 2015; 13(1):29-47. https://doi.org/10.3390/md13010029
Chicago/Turabian StyleXiao, Ai-Jiao, Wenliang Chen, Baofeng Xu, Rui Liu, Ekaterina Turlova, Andrew Barszczyk, Christopher Lf Sun, Ling Liu, Marielle Deurloo, Guan-Lei Wang, and et al. 2015. "Marine Compound Xyloketal B Reduces Neonatal Hypoxic-Ischemic Brain Injury" Marine Drugs 13, no. 1: 29-47. https://doi.org/10.3390/md13010029
APA StyleXiao, A. -J., Chen, W., Xu, B., Liu, R., Turlova, E., Barszczyk, A., Sun, C. L., Liu, L., Deurloo, M., Wang, G. -L., Feng, Z. -P., & Sun, H. -S. (2015). Marine Compound Xyloketal B Reduces Neonatal Hypoxic-Ischemic Brain Injury. Marine Drugs, 13(1), 29-47. https://doi.org/10.3390/md13010029