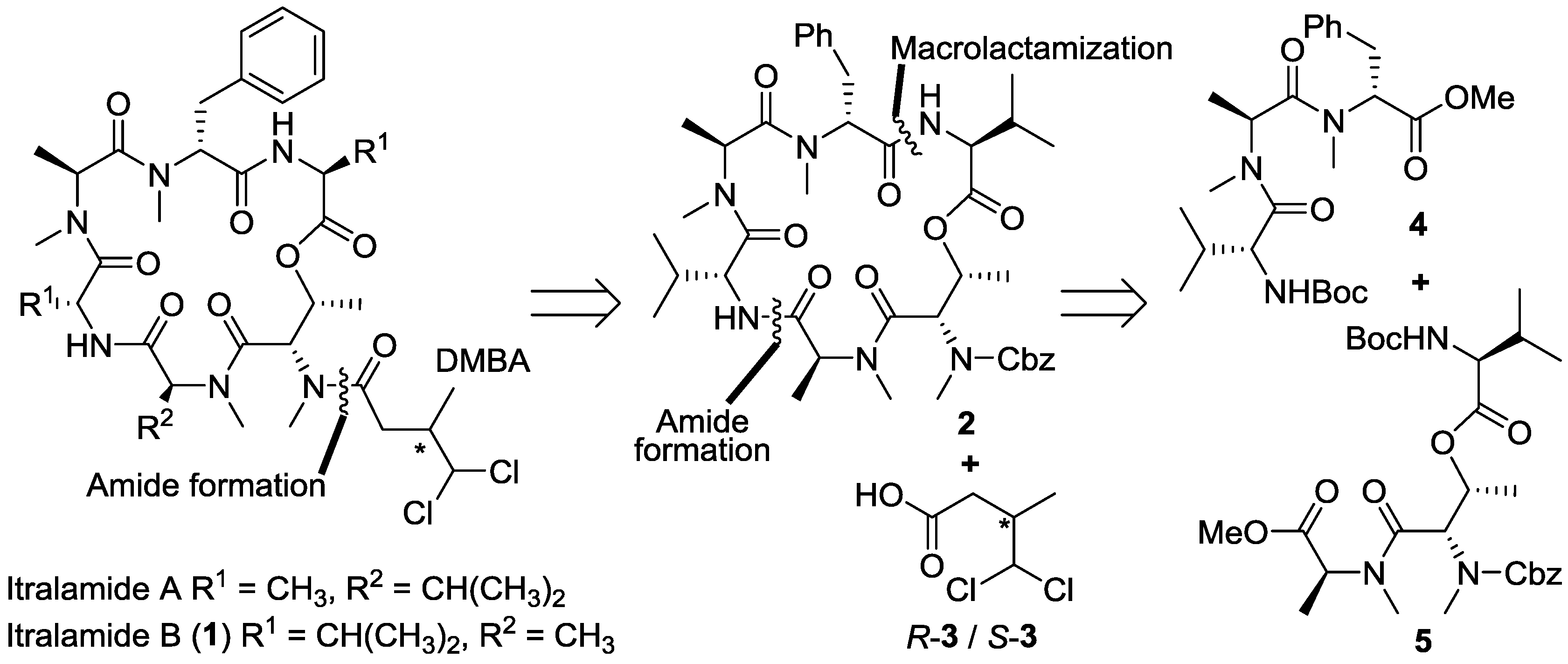
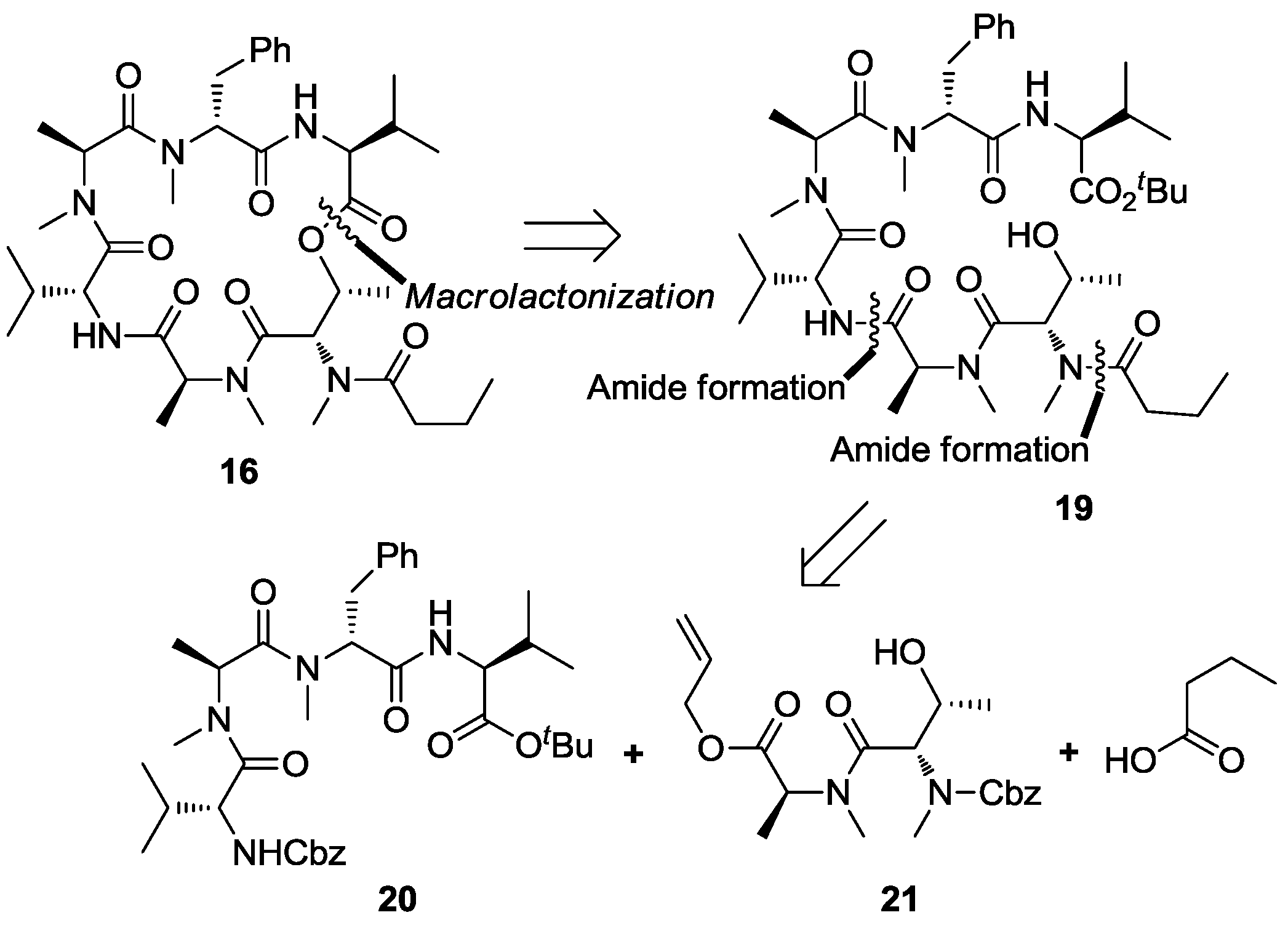
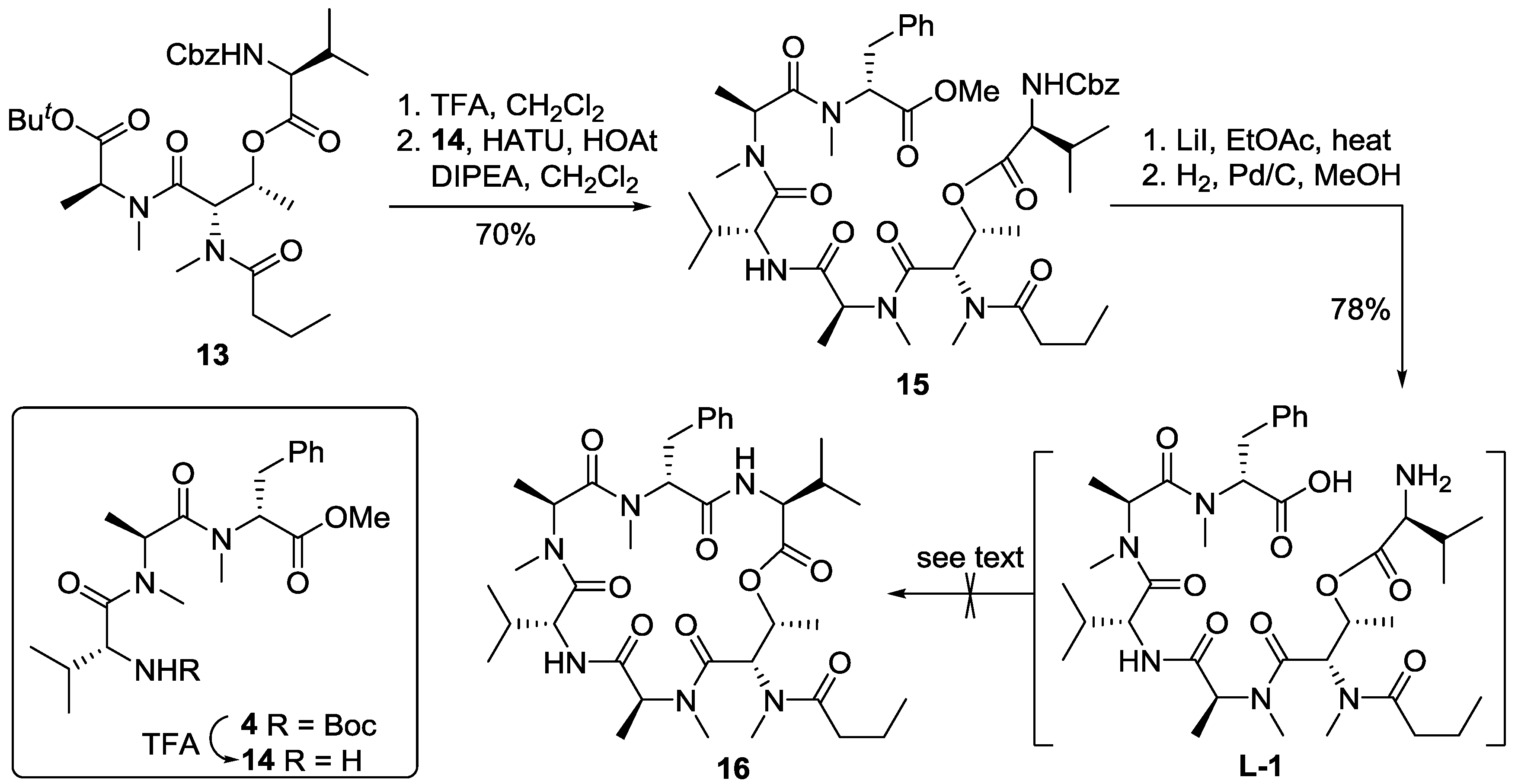
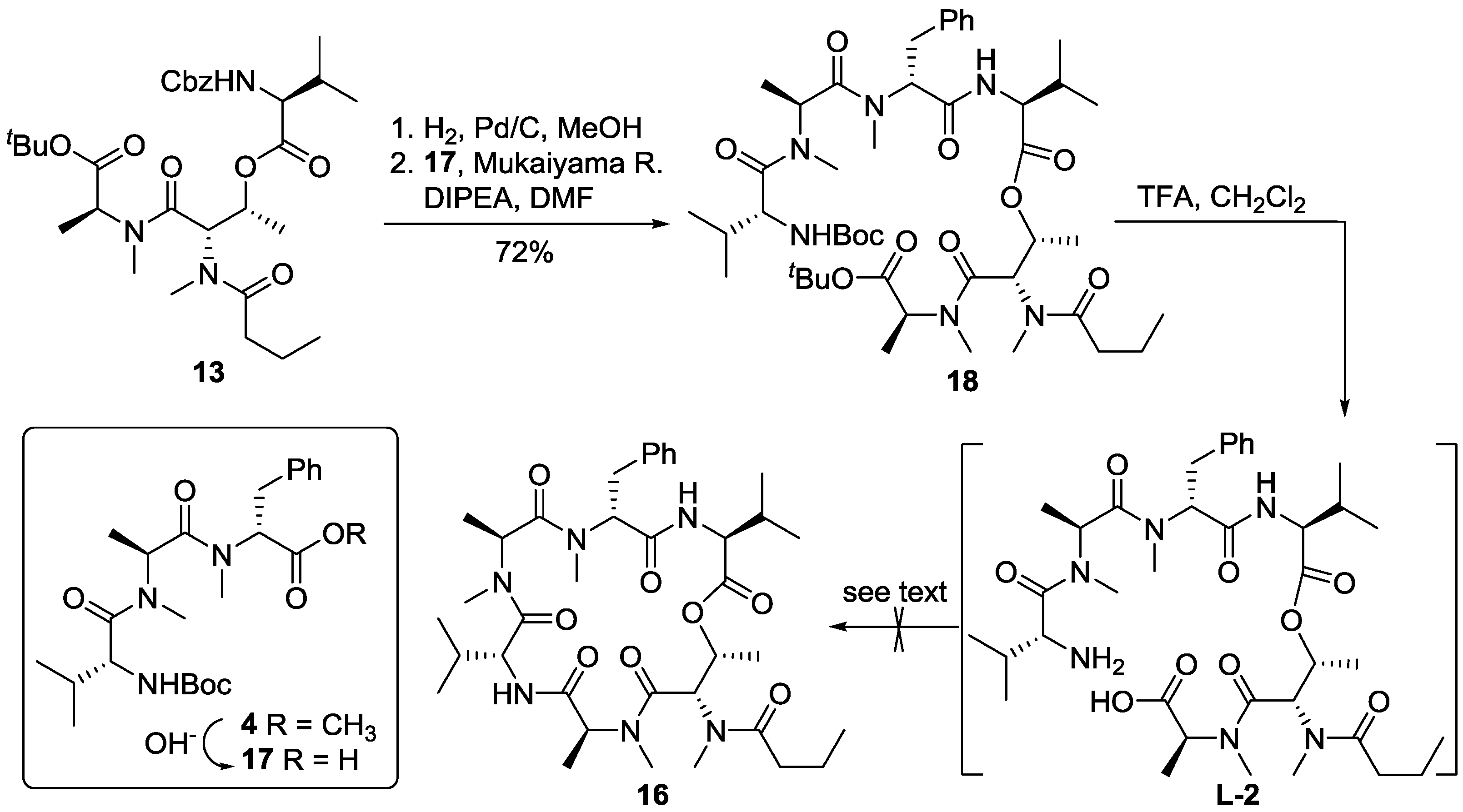
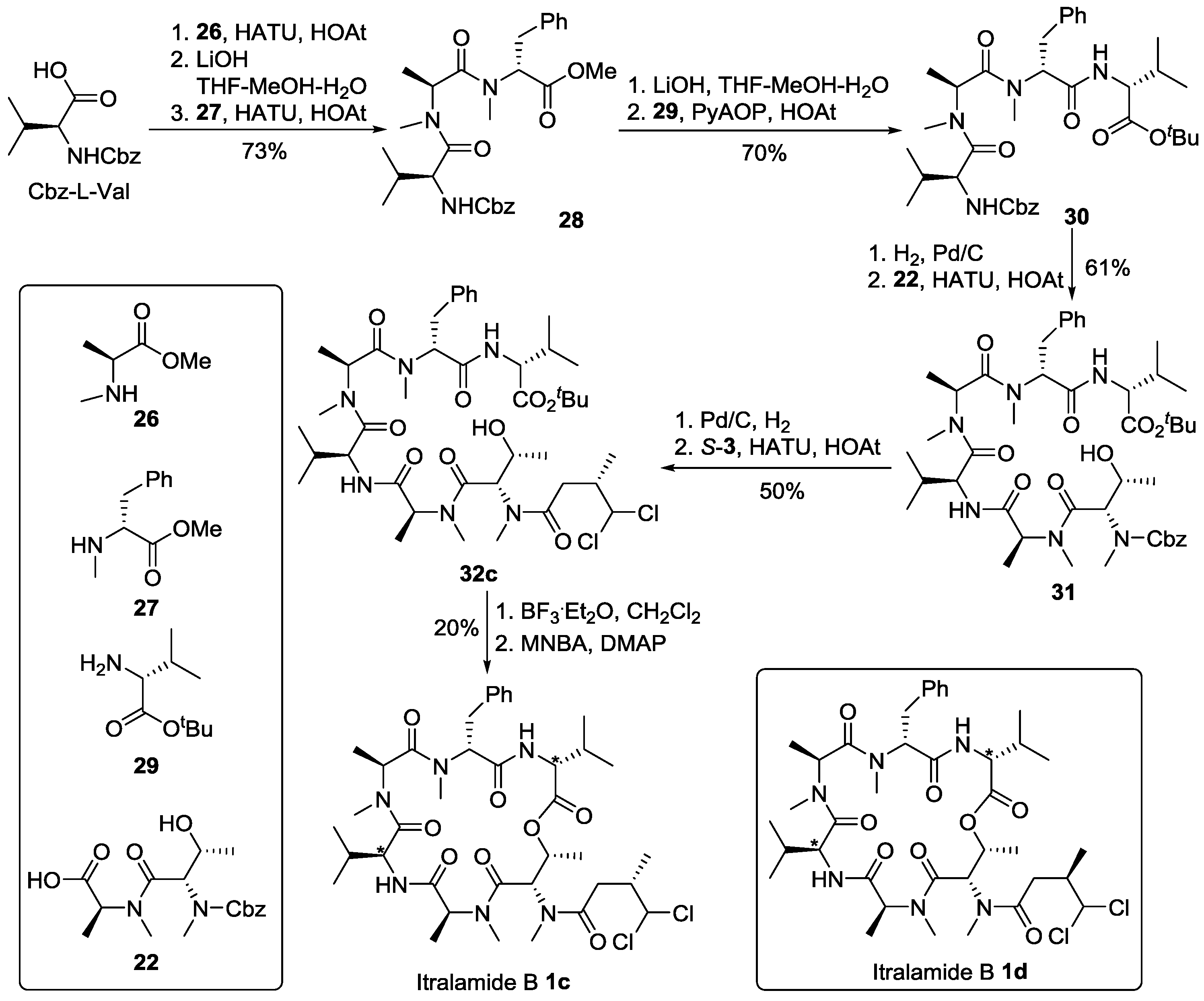
3.2. Synthesis of Cyclodepsipeptide 16 and Itralamide B 1c and 1d
3.2.1. Preparation of Cyclodepsipeptide 16
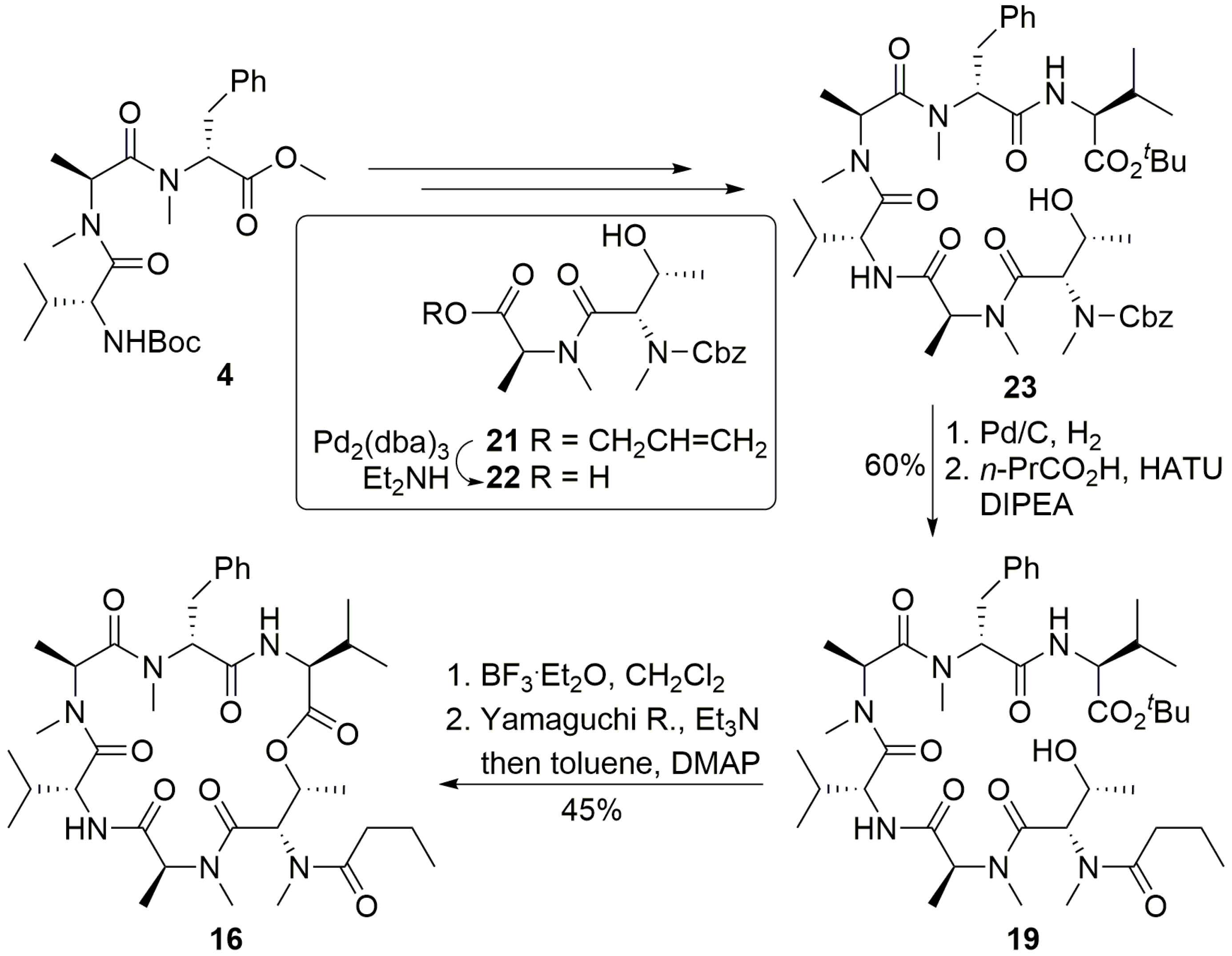
To a solution of compound 23 (200.0 mg, 0.23 mmol) in MeOH (20 mL), Pd/C was added under N2 atmosphere. The reaction vessel was sealed and flashed with H2 three times. The reaction mixture was then vigorously stirred overnight under H2 atmosphere. Catalyst residue was removed by filtration. The filtrate was concentrated in vacuo to give the corresponding free amine, which was pure enough and used directly in the next step of reaction. To a solution of n-PrCO2H (60.0 mg, 0.69 mmol) in DCM (2 mL), HATU (175.0 mg, 0.46 mmol) was added, followed by addition of DIPEA (200 μL, 1.15 mmol) at 0 °C. 0.5 h later, a solution of above amine (72.0 mg, 0.23 mmol) in DCM (2 mL) was addedat 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction was quenched with saturated NH4Cl (20 mL), and extracted with DCM (3 × 50 mL). The combined organic layers were washed with saturated NaHCO3 solution (3 × 50 mL) and brine (50 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (ethyl acetate) to give compound 19 (109.0 mg, 60%). [α]D25 = −45.3 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.25–7.15 (m, 5H), 6.71 (d, J = 8.9 Hz, 1H), 6.64 (d, J = 8.6 Hz, 1H), 5.43–5.41 (m, 1H), 5.17–5.08 (m, 1H), 4.66 (dd, J = 8.8, 6.3 Hz, 1H), 4.39 (dd, J = 6.5, 3.6 Hz, 1H), 4.34 (dd, J = 8.5, 4.8 Hz, 1H), 4.20 (d, J = 3.3 Hz, 1H), 3.37 (dd, J = 14.4, 7.4 Hz, 1H), 3.06 (s, 3H), 2.93 (s, 3H), 2.90–2.85 (m, 2H), 2.86 (s, 3H), 2.75 (s, 3H), 2.44–2.34 (m, 2H), 2.14–2.06 (m, 1H), 2.02–1.97 (m, 1H), 1.78−1.65 (m, 2H), 1.46 (s, 9H), 1.35 (d, J = 7.2 Hz, 3H), 1.15 (d, J = 6.4 Hz, 3H), 1.06 (d, J = 6.9 Hz, 3H), 0.99 (d, J = 5.1 Hz, 3H), 0.93 (d, J = 6.7 Hz, 3H), 0.87 (d, J = 6.8 Hz, 3H), 0.83 (d, J = 6.9 Hz, 3H), 0.82 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.7, 172.6, 172.0, 171.4, 170.8, 170.3, 169.4, 136.9, 129.0, 128.4, 126.6, 81.8, 68.1, 57.7, 56.8, 54.2, 52.5, 50.1, 38.6, 35.2, 34.0, 33.5, 31.0, 30.8, 30.7, 30.5, 28.0, 19.6, 18.9, 18.8, 18.5, 17.7, 17.4, 14.3, 13.9, 13.7. HR-ESIMS m/z for C41H68N6NaO9+ [M + Na]+: calculated 811.4940, found 811.4941.
To a solution of compound 19 (23.0 mg, 0.03 mmol) in DCM (1.0 mL), BF3.Et2O (38 μL, 0.3 mmol) was added dropwise at 0 °C. The reaction solution was then allowed to warm to room temperature and stirred for 0.5~1.0 h (monitored by TLC). The reaction was quenched by addition of saturated NH4Cl (2 mL) and diluted with DCM (60 mL). The organic phase was washed with saturated NH4Cl (3 × 20 mL) and brine (20 mL), dried over anhydrous Na2SO4 and concentrated in vacuo to produce crude hydroxy acid, which was dried further under high vacuum for 4 h. To a solution of the above acid (50.0 mg, 0.07 mmol) in THF (5 mL) was added Et3N (59 μL, 0.41 mmol) and trichlorobenzoyl chloride (54 μL, 0.34 mmol). The reaction mixture was stirred at room temperature for 3 h and diluted with toluene (3 mL). The resulted solution was added to a solution of DMAP (208.2 mg, 1.71 mmol) in toluene (50 mL) via a syringe pump over 48 h at 30 °C. The reaction was concentrated in vacuo, and the residue was dissolved in ethyl acetate (80 mL) and washed with saturated ammonium chloride (100 mL). Layers were separated, and the aqueous phase was extracted with ethyl acetate (2 × 80 mL). The combined organic layers were washed with brine (80 mL), dried over sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography (ethyl acetate) to give compound 16 (9.5 mg, 45%) as a yellow oil. [α]D25 = −65.4 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.24–7.16 (m, 5H), 6.89 (d, J = 9.6 Hz, 1H), 6.48 (d, J = 7.9 Hz, 1H), 5.77 (d, J = 3.2 Hz, 1H), 5.70 (dd, J = 12.3, 4.8 Hz, 1H), 5.47 (dd, J = 6.6, 3.2 Hz, 1H), 5.08 (q, J = 6.9 Hz, 1H), 4.98 (dd, J = 7.9, 4.3 Hz, 1H), 4.70 (dd, J = 9.4, 4.1 Hz, 1H), 4.66–4.58 (m, 1H), 3.81 (t, J = 7.8 Hz, 1H), 3.66 (dd, J = 15.3, 5.2 Hz, 1H), 3.33 (s, 3H), 3.19 (3.18) (s, 3H), 3.16 (s, 3H), 3.02 (s, 3H), 2.39–2.35 (m, 2H), 1.45–1.37 (m, 2H), 1.30 (d, J = 7.2 Hz, 3H), 1.07–1.04 (m, 3H), 0.99–0.79 (m, 18H). 13C NMR (100 MHz, CDCl3) δ 174.9, 172.9, 172.1, 170.7, 170.2, 170.0, 169.8, 137.4, 128.6, 128.3, 126.5, 69.8, 57.0, 56.8, 54.4, 54.0, 51.4, 50.3, 35.2, 33.8, 33.8, 32.2, 31.8, 31.1, 31.0, 30.5, 22.7, 19.9, 19.6, 18.6, 18.3, 17.8, 17.1, 17.1, 13.8. HR-ESIMS m/z calculated for C37H59N6O8+ [M + H]+: 715.4389, found 715.4390.
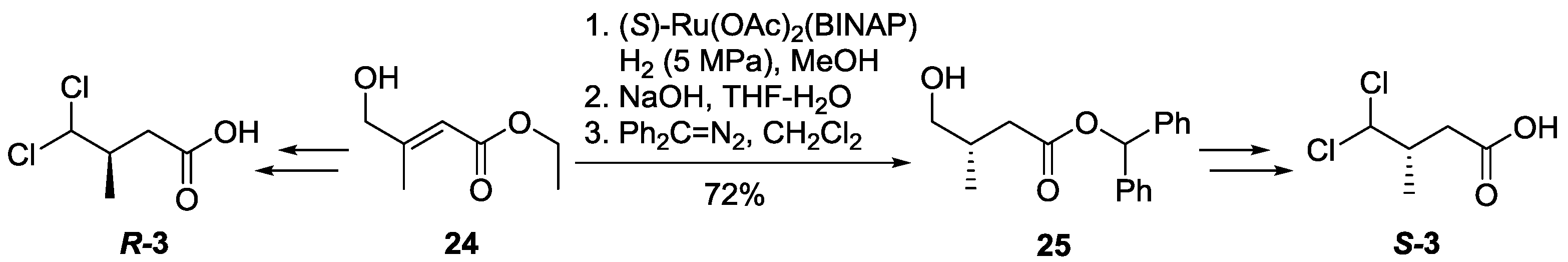
3.2.2. Preparation of Ester 25
In a stainless steel autoclave, ester
24 (461.4 mg, 3.19 mmol) was dissolved in methanol (50 mL), after catalyst (
S)-Ru(OAc)
2(BINAP) (48.0 mg, 0.06 mmol) was added, the reaction mixture was stirred under hydrogen atmosphere (5 MPa) for 24 h. The organic solution was transferred to a round bottom flask and concentrated to 5 mL, and THF–H
2O (10 mL, 1:1) was added, followed by addition of aqueous sodium hydroxide (6.4 mL, 6.4 mmol, 1 N in water). The solution was then stirred at room temperature for 12 h. Volatiles were removed under vacuum. The aqueous layer was extracted with diethyl ether (2 × 30 mL), and the organic solution was discarded. The aqueous solution was acidified to pH 3 with dilute hydrochloric acid (1 N in water) and extracted with dichloromethane (3 × 30 mL). The combined organic layers were dried over Na
2SO
4, filtered and concentrated in
vacuo to 10 mL. Without further purifications, to the above organic solution at 0 °C, diphenyldiazomethane (0.71 g, 3.66 mmol) in dichloromethane (3 mL) was added. The reaction mixure was stirred for an additional 6 h and then concentrated
in vacuo. The residue was purified using flash chromatography (ethyl acetate/hexane, 1:3) to provide
25 [
26] as a yellow oil (653.4 mg, 72%).
3.2.3. Preparation of Tripeptide 28
To a solution of Cbz-l-Val (9.20 g, 36.64 mmol) and amine 26 (4.31 g, 28.18 mmol) in DCM (250 mL), HATU (21.43 g, 56.36 mmol), DIPEA (23.3 mL, 140.90 mmol) and HOAt (7.67 g, 56.36 mmol) were added sequentially at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight, then quenched by addition of saturated NH4Cl solution (200 mL) and extracted with DCM (3 × 80 mL). The combined organic layers were washed with saturated NaHCO3 solution (3 × 80 mL) and brine (80 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (hexane/ethyl acetate, 1:1) to afford dipeptide Cbz-Val-MeAla-OMe (6.27 g, 64%). [α]D20 = −21.5, (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.40–7.25 (m, 5H), 5.59 (d, J = 7.5 Hz, 1H), 5.27 (q, J = 7.1 Hz, 1H), 5.15–5.04 (m, 2H), 4.55 (dd, J = 9.2, 5.9 Hz, 1H), 3.70 (s, 3H), 3.03 (2.84) (s, 3H), 2.10–2.00 (m, 1H), 1.41 (d, J = 7.4 Hz, 3H), 1.03 (d, J = 6.7 Hz, 3H), 1.00–0.87 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 172.3, 172.0, 156.5, 136.4, 128.5, 128.1, 128.0, 66.9, 55.8, 52.2, 52.1, 31.3, 31.3, 19.4, 17.2, 14.1.
To a solution of Cbz-Val-MeAla-OMe (4.27 g, 12.19 mmol) in THF-MeOH-H2O (90 mL, 1:1:1) was added LiOH.H2O (1.46 g, 60.93 mmol) at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for 5 h (monitored by TLC). Volatiles were removed in vacuo, the aqueous solution was washed with Et2O (3 × 80 mL). The organic phases were discarded, and the aqueous phase was acidified to pH 3 with 10% aqueous solution of citric acid at 0 °C. The aqueous layer was extracted with ethyl acetate (3 × 80 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give the corresponding acid (4.10 g, 99%). This acid (4.10 g, 12.18 mmol), without further purification, was mixed with amine 27 (3.63 g, 15.84 mmol) and dissolved in DCM (80 mL) at 0 °C. To this solution, HATU (9.27 g, 24.38 mmol), DIPEA (10.1 mL, 60.95 mmol) and HOAt (3.32 g, 24.38 mmol) were added at 0 °C. The reaction mixture was then allowed to warm to room temperature and stirred for 16 h. The reaction was quenched with saturated NH4Cl solution (200 mL). Layers were separated, the aqueous layer was extracted with DCM (3 × 80 mL). The combined organic layers were washed with saturated NaHCO3 solution (3 × 80 mL) and brine (80 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (hexane/ethyl acetate, 1:1) to afford tripeptide 28 (4.36 g, 70%) as a viscous oil. [α]D25 = +85.8 (c 2.3, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.39–7.25 (m, 7H), 7.24–7.10 (m, 3H), 5.59–5.45 (m, 1H), 5.43–5.13 (m, 2H), 5.12–4.82 (m, 2H), 4.51–4.25 (m, 1H), 3.73 (3.54) (s, 3H), 3.45–3.25 (m, 1H), 2.95–2.87 (m, 1H), 2.85 (2.82) (s, 3H), 2.61 (2.29) (s, 3H), 2.00–1.85 (m, 1H), 1.25–0.82 (m, 9H). 13C NMR (125 MHz, CDCl3) δ 171.7, 171.5, 171.0, 170.9, 170.8, 170.5, 170.4, 156.2, 136.9, 136.8, 136.4, 129.0, 129.0, 128.8, 128.7, 128.5, 128.1, 128.0, 128.0, 127.9, 127.0, 126.8, 66.9, 66.8, 66.8, 60.4, 60.4, 59.0, 58.3, 55.9, 55.6, 55.5, 52.5, 52.3, 49.8, 48.4, 35.1, 34.5, 34.4, 32.3, 32.3, 31.6, 31.2, 31.0, 30.0, 29.9, 29.3, 21.0, 19.8, 19.4, 19.2, 17.6, 17.1, 16.8, 14.3, 14.2, 14.0; HR-ESIMS m/z for C28H38N3O6+ [M + H]+: calculated 512.2755, found 512.2755.
3.2.4. Preparation of Tetrapeptide 30
To a solution of the tripeptide 28 (1.00 g, 1.95 mmol) in THF–MeOH–H2O (30 mL, 1:1:1) was added LiOH.H2O (250.0 mg, 5.94 mmol) at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for 5 h (monitored by TLC). Volatiles were removed in vacuo. The aqueous layer was washed with Et2O (3 × 80 mL). The organic phases were discarded, and the aqueous phase was acidified to pH 3 with 10% aqueous solution of citric acid at 0 °C. This aqueous layer was then extracted with ethyl acetate (3 × 80 mL). The combined organic layers were washed with brine (50 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give the corresponding acid (850.0 mg, 87%). To a solution of above acid in DCM (50 mL), PyAOP (1.78 g, 3.42 mmol), DIPEA (1.4 mL, 8.55 mmol) and HOAt (470.0 mg, 3.42 mmol) were sequentially added at 0 °C. 0.5 h later, a solution of Val-OtBu 29 (360.0 mg, 2.05 mmol) in DCM (5 mL) was added at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for 16 h. The reaction was quenched by addition of saturated NH4Cl solution (150 mL). Layers were separated, the aqueous phase was extracted with DCM (3 × 80 mL). The combined organic layers were washed with saturated NaHCO3 solution (3 × 50 mL) and brine (30 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (hexane/ethyl acetate, 1:1) to afford tetrapepetide 30 (890.0 mg, 80%). [α]D25 = +32.5 (c 1.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.39–7.11 (m, 10H), 6.47–6.12 (m, 1H), 5.53–5.41 (m, 1H), 5.41–5.35 (m, 1H), 5.22–5.06 (m, 2H), 4.96–4.92 (m, 1H), 4.45–4.40 (m, 1H), 4.33–4.28 (m, 1H), 3.26–3.20 (m, 1H), 3.10 (2.74) (s, 3H), 2.95–2.88 (m, 1H), 2.74 (2.33) (s, 3H), 2.17–2.09 (m, 1H), 1.96–1.85 (m, 1H), 1.45 (1.32) (s, 9H), 1.11–0.90 (m, 15H). 13C NMR (125 MHz, CDCl3) δ 172.2, 171.4, 170.9, 170.5, 170.0, 168.8, 156.5, 156.2, 137.7, 137.1, 136.6, 136.6, 129.6, 129.1, 128.9, 128.7, 128.6, 128.3, 128.3, 128.2, 128.0, 127.9, 127.0, 126.7, 82.1, 81.7, 66.9, 62.0, 58.2, 57.7, 56.8, 55.5, 50.0, 47.2, 34.9, 33.3, 31.7, 31.4, 31.4, 30.8, 30.7, 29.3, 28.9, 28.0, 27.8, 19.5, 19.3, 19.0, 18.8, 17.9, 17.6, 17.5, 17.5, 14.3, 14.2. HR-ESIMS m/z for C36H53N4O7+ [M + H]+: calculated 653.3909, found 653.3908.
3.2.5. Preparation of Hexapeptide 31
To a solution of tetrapeptide 30 (620.0 mg, 0.95 mmol) in MeOH (30 mL), Pd/C was added under N2 atmosphere. The reaction vessel was sealed and flashed with H2 for three times. The reaction mixture was then vigirously stirred overnight under a H2 atmosphere. Catalyst was removed by filtration. The filtrate was concentrated in vacuo to give the corresponding free amine, which was pure enough and used directly in next step of reaction. To a solution of dipeptide 22 (220.0 mg, 0.43 mmol) in DCM (15 mL) was added HATU (323.0 mg, 0.85 mmol), followed by addition of DIPEA (0.4 mL, 2.13 mmol) and HOAt (116.0 mg, 0.85 mmol) at 0 °C. 0.5 h later, a solution of the above free amine in DCM (5 mL) was added at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight, then quenched by addition of saturated NH4Cl (40 mL). Layers were seperated, the aqueous phase was extracted with DCM (3 × 80 mL). The combined organic layers were washed with saturated NaHCO3 solution (3 × 50 mL) and brine (30 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (hexane/ethyl acetate, 1:1) to afford 31 (221.0 mg, 61%). [α]D25 = +8.3 (c 0.8, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.43–7.01 (m, 11H), 6.64 (6.46) (d, J = 8.8 Hz, 1H), 5.49–5.34 (m, 1H), 5.33–5.17 (m, 1H), 5.17–4.95 (m, 2H), 4.85–4.80 (m, 1H), 4.78–4.43 (m, 2H), 4.43–4.18 (m, 2H), 3.27 (dd, J = 14.6, 5.6 Hz, 1 H), 3.12 (s, 1H), 3.10–3.01 (m, 3H), 2.97–2.93 (m, 3H), 2.90–2.83 (m, 3H), 2.80–2.77 (m, 2H), 2.31 (s, 2H), 2.26–2.09 (m, 1H), 2.01–1.85 (m, 1H), 1.80 (s, 1H), 1.41 (1.40) (s, 9H), 1.30–0.70 (m, 20 H), 0.46–0.27 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 172.4, 172.3, 171.8, 171.4, 170.7, 170.4, 169.8, 168.8, 137.6, 137.0, 136.3, 129.6, 128.9, 128.9, 128.6, 128.6, 128.5, 128.1, 127.7, 126.9, 126.7, 81.7, 81.7, 67.8, 67.7, 62.1, 59.3, 58.0, 57.6, 54.3, 53.3, 52.4, 50.0, 47.7, 33.2, 32.1, 32.0, 31.5, 31.1, 31.0, 30.9, 30.8, 30.7, 30.4, 29.3, 28.9, 27.9, 19.4, 18.9, 18.5, 18.2, 18.0, 17.4, 17.2, 14.5, 14.3, 14.1, 13.6. HR-ESIMS m/z for C45H68N6NaO10+ [M + Na]+: calculated 875.4889, found 875.4891.
3.2.6. Preparation of 32c and 32d
To a solution of hexapeptide 31 (40.0 mg, 0.05 mmol) in MeOH (20 mL), was added Pd/C (10% on charcoal) under N2 atmosphere. The reaction vessel was sealed and flashed with H2 for three times. The reaction mixture was then vigirously stirred overnight under H2 atmosphere. Catalyst was removed by filtration. The filtrate was concentrated in vacuo to give the corresponding free amine, which was pure enough and used directly in next step of reaction. To a solution of S-3 (23.0 mg, 0.14 mmol) in DCM (2 mL) was added HATU (34.0 mg, 0.09 mmol), followed by addition of DIPEA (39 μL, 0.23 mmol) and HOAt (12.0 mg, 0.09 mmol) at 0 °C. 0.5 h later, a solution of the above amine (32.0 mg, 0.04 mmol) in DCM (2 mL) was added at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction was quenched with saturated NH4Cl (20 mL), and extracted with DCM (3 × 50 mL). The combined organic layers were washed with saturated NaHCO3 solution (3 × 50 mL) and brine (50 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (ethyl acetate) to give compound 32c (20.0 mg, 50%). [α]D25 = −12.3 (c 0.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 9.0 Hz, 0.5H), 7.32–7.28 (m, 1H), 7.26–7.14 (m, 4H), 6.96 (d, J = 9.5 Hz, 0.4H), 6.68 (d, J = 8.9 Hz, 0.4H), 6.50 (d, J = 8.9 Hz, 0.6H), 6.08–6.01 (m, 1H), 5.52–5.37 (m, 1H), 5.36–5.30 (m, 1H), 5.14–5.01 (m, 1H), 4.91–4.65 (m, 1H), 4.59–4.42 (m, 1H), 4.30 (dd, J = 8.8, 4.6 Hz, 2H), 3.31–3.18 (m, 1H), 3.17–3.08 (m, 1H), 3.07–2.82 (m, 9H), 2.80–2.74 (m, 3H), 2.51–2.37 (m, 1H), 2.37–2.29 (m, 2H), 2.28–2.17 (m, 1H), 2.17–2.07 (m, 1H), 2.02–1.84 (m, 2H), 1.43 (s, 9H), 1.39–1.28 (m, 3H), 1.19–1.07 (m, 6H), 1.05–0.97 (m, 3H), 0.91–0.76 (m, 12H). 13C NMR (75 MHz, CDCl3) δ 172.5, 172.4, 172.3, 171.9, 171.8, 171.4, 170.7, 170.4, 169.9, 168.8, 156.8, 137.6, 137.0, 136.3, 129.6, 128.9, 128.9, 128.6, 128.5, 128.1, 127.7, 126.9, 126.7, 81.7, 68.5, 68.2, 68.0, 67.8, 62.9, 62.1, 60.3, 59.3, 58.9, 58.2, 58.0, 57.9, 57.6, 56.9, 54.3, 53.3, 52.4, 50.9, 50.0, 47.7, 47.5, 43.2, 34.7, 33.2, 32.1, 32.0, 31.5, 31.1, 31.0, 30.7, 30.4, 29.3, 28.9, 27.9, 22.4, 19.4, 19.4, 18.9, 18.5, 18.2, 18.0, 17.4, 17.2, 15.0, 14.5, 14.3, 14.1, 13.6. HR-ESIMS m/z for C42H68Cl2N6NaO9+ [M + Na]+: calculated 893.4317, found 893.4318.
Compound 32d was prepared in 52% yield. Analytical data: [α]D25 = −28.2 (c 0.3, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.09 (d, J = 9.0 Hz, 0.5H), 7.43–7.13 (m, 5H), 7.00 (d, J = 9.5 Hz, 0.5H), 6.70 (d, J = 8.9 Hz, 0.5H), 6.52 (d, J = 8.9 Hz, 0.5H), 6.15–5.91 (m, 1H), 5.58–5.38 (m, 1H), 5.38–5.23 (m, 1H), 5.16–5.01 (m, 1H), 4.93–4.65 (m, 1H), 4.54 (ddd, J = 14.3, 8.9, 5.8 Hz, 1H), 4.32 (dd, J = 8.8, 4.6 Hz, 2H), 3.26 (dt, J = 16.8, 7.2 Hz, 1H), 3.17–3.07 (m, 3H), 3.07–2.90 (m, 3H), 2.90 (2.88) (s, 3H), 2.86–2.76 (m, 3H), 2.71 (dt, J = 16.7, 6.2 Hz, 1H), 2.55–2.42 (m, 1H), 2.32 (s, 1H), 2.17 (ddd, J = 14.0, 12.4, 6.6 Hz, 1H), 2.03–1.87 (m, 1H), 1.46 (s, 9H), 1.39–1.35 (m, 3H), 1.26–1.22 (m, 6H), 1.15–1.11 (m, 3H), 1.04 (t, J = 6.5 Hz, 3H), 0.93–0.77 (m, 9H), 0.38 (d, J = 6.9 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 172.5, 172.4, 172.3, 172.0, 171.9, 171.4, 171.2, 171.0, 170.7, 170.6, 170.4, 169.9, 169.8, 168.9, 137.6, 137.0, 129.6, 129.0, 128.9, 128.7, 127.0, 126.7, 84.6, 81.7, 68.6, 62.0, 58.0, 57.9, 57.6, 57.3, 57.0, 55.7, 54.3, 52.6, 52.4, 51.7, 50.0, 48.8, 47.8, 45.6, 40.3, 35.6, 35.6, 34.7, 33.3, 31.5, 31.2, 31.0, 30.9, 30.7, 30.7, 30.6, 30.5, 29.6, 29.3, 28.9, 27.9, 19.4, 19.4, 18.9, 18.7, 18.5, 18.0, 17.4, 17.3, 15.2, 14.6, 14.3, 14.1, 13.7. HR-ESIMS m/z for C42H68Cl2N6NaO9+ [M + Na]+: calculated 893.4317, found 893.4316.
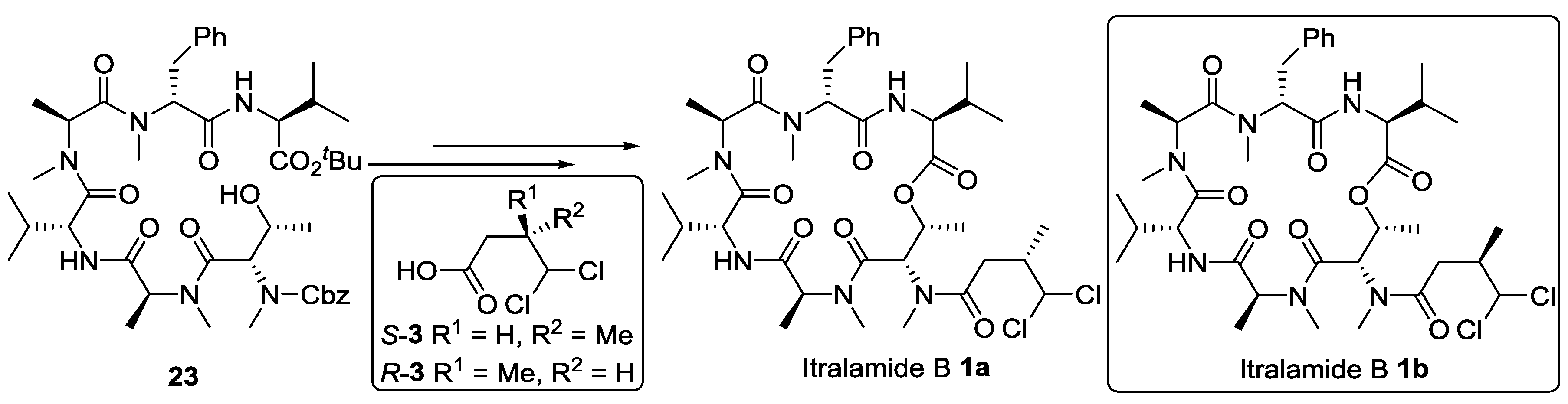
3.2.7. Completion of the Synthesis of Itralamide B 1c and 1d
To a solution of compound 32c (15.0 mg, 0.02 mmol) in DCM (1.0 mL), BF3.Et2O (21 μL, 0.17 mmol) was added dropwise at 0 °C. The reaction solution was then allowed to warm to room temperature and stirred for 0.5~1.0 h (monitored by TLC). The reaction was quenched by addition of saturated NH4Cl (2 mL) and diluted with DCM (60 mL). The organic phase was washed with saturated NH4Cl (3 × 20 mL) and brine (20 mL), dried over anhydrous Na2SO4 and concentrated in vacuo to give crude hydroxy acid, which was dried under high vacuum for 4 h. To a solution of DMAP (21.0 mg, 0.17 mmol) and MNBA (30.0 mg, 0.08 mmol), a solution of above hydroxy acid in toluene (5 mL) was slowly added at 0 °C. After the reaction mixture was warmed to room temperature, it was gradually heated to 60 °C and stirred for two days. The reaction mixture was diluted with ethyl acetate (100 mL) and washed successively with saturated NH4Cl (3 × 20 mL) and brine (2 × 20mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (ethyl acetate) to afford itralamide B 1c (2.7 mg, 20%). [α]D25 = −10.8 (c 0.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.45 (d, J = 7.6 Hz, 1H), 7.33–7.28 (m, 2H), 7.19 (d, J = 7.0 Hz, 3H), 5.98 (d, J = 3.0 Hz, 1H), 5.80 (d, J = 3.3 Hz, 1H), 5.64 (dd, J = 6.7, 3.1 Hz, 1H), 5.34 (dd, J = 11.4, 3.7 Hz, 1H), 5.12 (q, J = 6.9 Hz, 1H), 4.98 (q, J = 6.7 Hz, 1H), 4.67 (t, J = 10.1 Hz, 1H), 4.63 (dd, J = 7.5, 3.8 Hz, 1H), 3.36 (s, 3H), 3.23–3.10 (m, 2H), 3.08 (s, 3H), 3.05 (s, 3H), 2.99 (s, 3H), 2.91 (dd, J = 14.3, 3.4 Hz, 2H), 2.85 (d, J = 8.9 Hz, 1H), 2.81–2.74 (m, 2H), 2.70 (dd, J = 16.6, 5.1 Hz, 2H), 2.47 (dd, J = 16.7, 7.6 Hz, 1H), 2.32–2.27 (m, 1H), 2.27–2.21 (m, 1H), 2.02 (s, 1H), 1.36 (d, J = 6.7 Hz, 3H), 1.20 (d, J = 6.6 Hz, 3H), 1.05 (d, J = 6.9 Hz, 3H), 0.97 (d, J = 6.7 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H), 0.90 (s, 1H), 0.88 (d, J = 6.8 Hz, 3H), 0.41 (d, J = 6.8 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 172.4, 172.2, 171.4, 170.2, 169.5, 168.8, 137.2, 132.1, 132.0, 131.9, 129.7, 128.9, 128.5, 128.4, 127.0, 78.0, 69.8, 61.2, 57.6, 55.5, 53.5, 52.7, 46.6, 40.6, 35.7, 35.7, 34.1, 32.5, 31.9, 31.4, 30.7, 29.8, 29.7, 29.6, 29.4, 28.8, 22.7, 19.6, 18.8, 18.7, 18.0, 17.6, 15.4. HR-ESIMS m/z for C38H58Cl2N6NaO8+ [M + Na]+: calculated 819.3585, found 819.3587.
Compound itralamide B 1d was prepared in 21% yield. Analytical data: [α]D25 = −8.3 (c 0.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.46 (d, J = 7.5 Hz, 1H), 7.32 (dd, J = 9.6, 5.4 Hz, 2H), 7.20 (d, J = 7.1 Hz, 3H), 6.00 (d, J = 2.9 Hz, 1H), 5.82 (d, J = 3.1 Hz, 1H), 5.66 (dd, J = 6.7, 3.2 Hz, 1H), 5.41–5.29 (m, 2H), 5.14 (q, J = 6.9 Hz, 1H), 5.00 (q, J = 6.8 Hz, 1H), 4.69 (t, J = 10.1 Hz, 1H), 4.64 (dd, J = 7.6, 3.8 Hz, 1H), 3.38 (s, 3H), 3.19 (dd, J = 14.0, 11.5 Hz, 1H), 3.10 (s, 3H), 3.07 (s, 3H), 3.01 (s, 3H), 2.98–2.94 (m, 2H), 2.94–2.90 (m, 1H), 2.80 (d, J = 18.3 Hz, 2H), 2.72 (dd, J = 16.6, 5.1 Hz, 1H), 2.49 (dd, J = 16.7, 7.6 Hz, 1H), 2.38 (d, J = 6.4 Hz, 1H), 2.34–2.30 (m, 1H), 2.25 (dd, J = 9.7, 5.7 Hz, 1H), 2.03 (s, 1H), 1.38 (d, J = 6.7 Hz, 3H), 1.22 (d, J = 6.6 Hz, 3H), 1.07 (d, J = 7.0 Hz, 3H), 0.99 (d, J = 6.7 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.92 (s, 1H), 0.90 (d, J = 6.7 Hz, 3H), 0.43 (d, J = 6.8 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 172.8, 172.4, 172.2, 171.4, 170.2, 169.5, 168.8, 137.2, 129.7, 128.9, 127.0, 78.0, 69.8, 61.2, 57.6, 55.5, 53.5, 52.7, 46.6, 40.6, 35.9, 35.7, 34.2, 32.5, 31.9, 31.9, 31.4, 30.7, 29.8, 29.7, 29.3, 28.8, 22.7, 19.6, 18.8, 18.7, 18.0, 17.6, 15.4, 14.2, 14.1, 13.4. HR-ESIMS m/z for C38H58Cl2N6NaO8+ [M + Na]+: calculated 819.3585, found 819.3589.
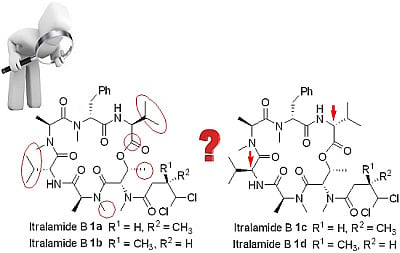
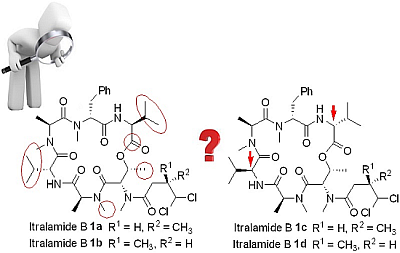
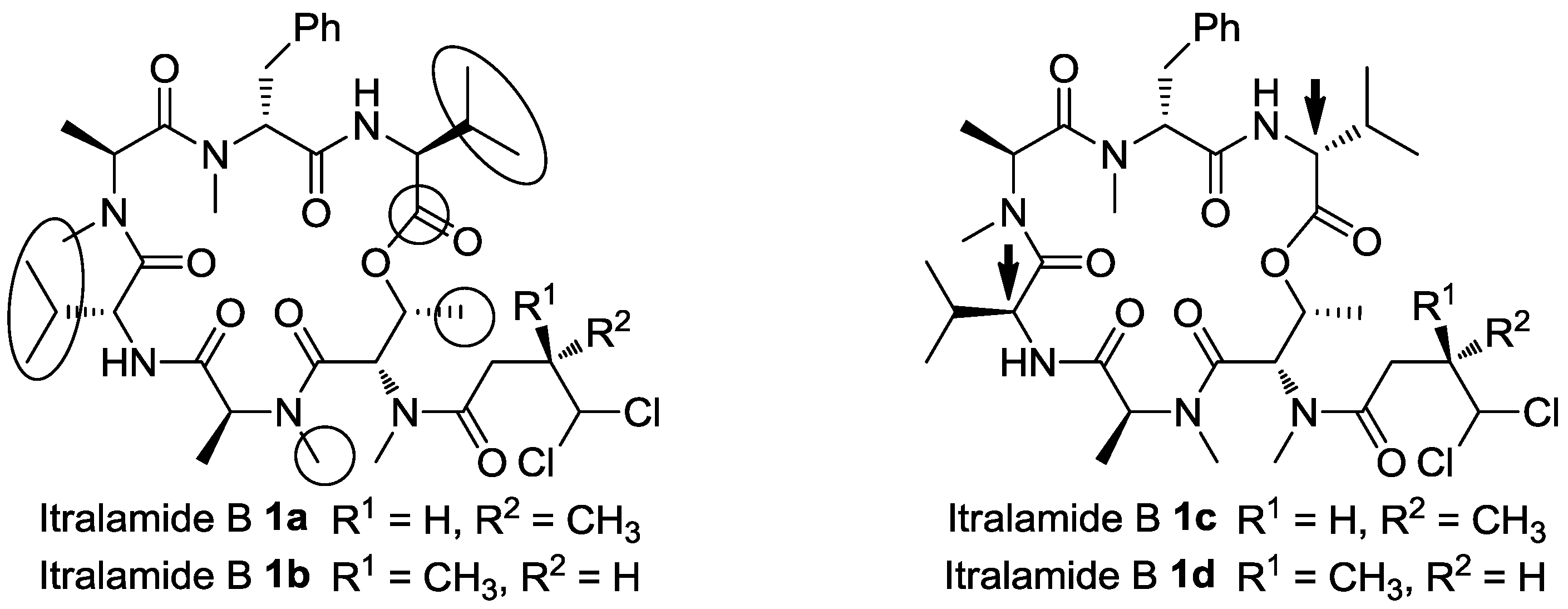
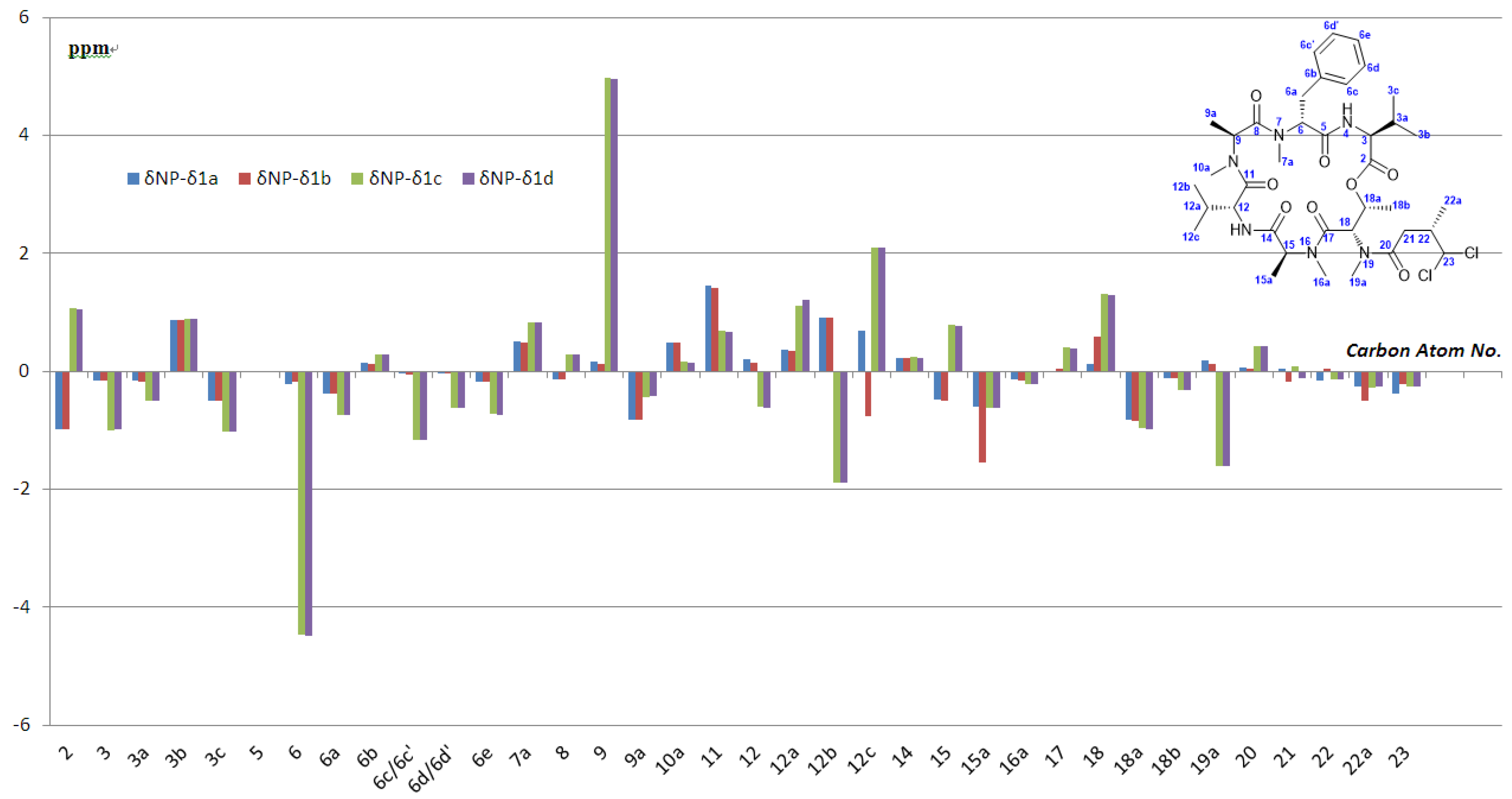
3.2.8. Analytical Data of Itralamide B 1a and 1b [26]
Itralamide B 1a: [α]D25 = −50.9 (c 0.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.45–7.12 (m, 5H), 6.90 (d, J = 9.6 Hz, 1H), 6.47 (d, J = 8.0 Hz, 1H), 5.99 (d, J = 2.9 Hz, 1H), 5.75–5.65 (m, 2H), 5.53–5.43 (m, 1H), 5.08 (q, J = 6.8 Hz, 1H), 4.98 (dd, J = 7.9, 4.2 Hz, 1H), 4.71 (dd, J = 9.4, 4.1 Hz, 1H), 4.62 (q, J = 7.6 Hz, 1H), 3.66 (dd, J = 15.5, 4.9 Hz, 1H), 3.33 (d, J = 23.6 Hz, 3H), 3.19 (d, J = 7.1 Hz, 3H), 3.17–3.07 (m, 3H), 3.07–2.94 (m, 3H), 2.87 (dd, J = 15.6, 12.1 Hz, 1H), 2.80 (d, J = 4.0 Hz, 1H), 2.76–2.65 (m, 1H), 2.46 (dd, J = 16.5, 7.5 Hz, 1H), 2.28–2.21 (m, 1H), 2.06–1.99 (m, 1H), 1.29 (d, J = 2.0 Hz, 3H), 1.26 (d, J = 2.8 Hz, 3H), 1.20 (dd, J = 6.6, 3.5 Hz, 3H), 1.08–1.01 (m, 6H), 0.92 (d, J = 7.2 Hz, 6H), 0.80 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.9, 172.8, 172.5, 170.6, 170.2, 169.9, 169.5, 137.4, 128.5, 128.3, 126.5, 78.2, 69.6, 56.9, 56.8, 54.7, 54.7, 54.0, 51.4, 40.6, 35.8, 33.9, 33.8, 32.2, 31.8, 31.1, 31.0, 30.5, 19.9, 19.6, 17.8, 17.1, 15.6, 15.4, 14.1, 13.8. HR-ESIMS m/z for C38H58Cl2N6NaO8+ [M + Na]+: calculated 819.3585, found 819.3587.
Itralamide B 1b: [α]D25 = −42.3 (c 0.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.24–7.16 (m, 5H), 6.89 (d, J = 9.1 Hz, 1H), 6.48 (d, J = 7.8 Hz, 1H), 6.04 (d, J = 2.9 Hz, 1H), 5.78–5.63 (m, 1H), 5.49 (dd, J = 6.6, 3.2 Hz, 1H), 5.45–5.35 (m, 1H), 5.12–5.02 (m, 1H), 4.98 (dd, J = 7.8, 4.2 Hz, 1H), 4.71 (dd, J = 9.3, 4.0 Hz, 1H), 4.67–4.55 (m, 1H), 3.66 (dd, J = 15.3, 5.1 Hz, 1H), 3.28 (d, J = 63.9 Hz, 3H), 3.18 (s, 3H), 3.13 (d, J = 23.1 Hz, 3H), 3.01 (d, J = 11.3 Hz, 3H), 2.93 (d, J = 15.7 Hz, 1H), 2.90–2.78 (m, 1H), 2.78–2.67 (m, 1H), 2.43 (dd, J = 16.7, 6.0 Hz, 1H), 2.25 (dd, J = 11.9, 5.9 Hz, 1H), 2.04 (d, J = 9.5 Hz, 1H), 1.38 (d, J = 7.2 Hz, 3H), 1.30 (d, J = 6.7 Hz, 3H), 1.20 (d, J = 6.6 Hz, 3H), 1.06 (d, J = 6.8 Hz, 6H), 0.92 (d, J = 7.0 Hz, 6H), 0.80 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.9, 172.8, 172.6, 170.7, 170.2, 169.8, 169.5, 137.4, 128.6, 128.3, 126.5, 78.0, 69.6, 56.9, 56.8, 54.8, 54.2, 54.0, 51.5, 40.4, 36.0, 34.0, 33.8, 32.2, 31.8, 31.1, 31.0, 30.6, 19.9, 19.6, 17.8, 17.1, 17.1, 15.6, 15.0, 13.8. HR-ESIMS m/z for C38H58Cl2N6NaO8+ [M + Na]+: calculated 819.3585, found 819.3589.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}