
Esters of the Marine-Derived Triterpene Sipholenol A Reverse P-GP-Mediated Drug Resistance


 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell lines and Cell Culture
2.3. Cell Cytotoxicity by MTT Assay
2.4. Preparation of Total Cell Lysates
2.5. Western Blot Analysis
2.6. [3H]-Paclitaxel Accumulation and Efflux Assays
2.7. ATPase Assay of SSJ26 and SSJ32
2.8. Molecular Docking Analysis
2.9. Statistical Analysis
3. Results
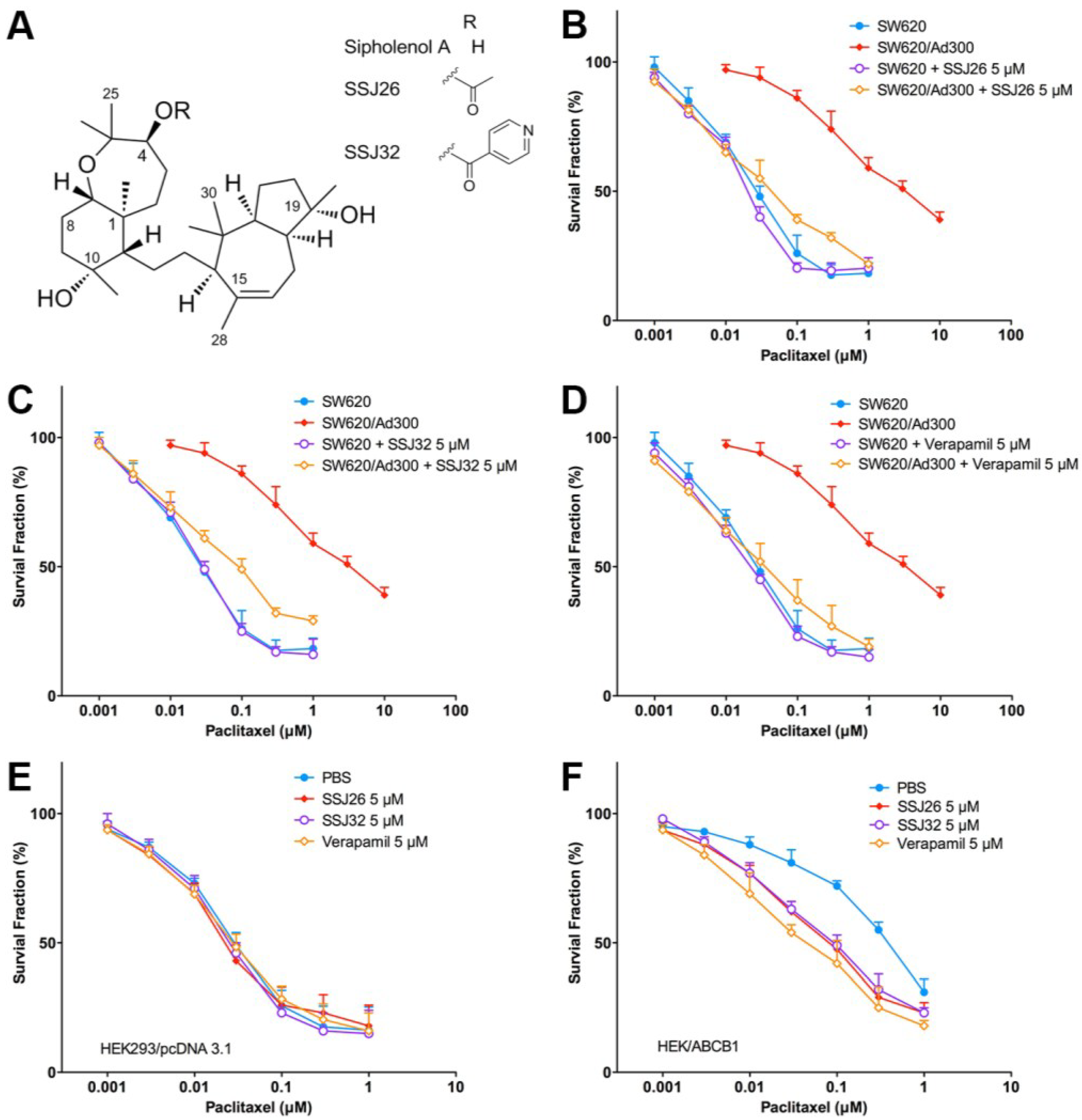
3.1. Screening for Potential Inhibitors of P-GP Transporter in Comparison with Verapamil

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | SW620 | SW620/Ad300 | ||
|---|---|---|---|---|
| IC50 ± SD a (μM) | FR b | IC50 ± SD a (μM) | FR b | |
| Doxorubicin | 0.0196 ± 0.0022 | 1.00 | 4.1082 ± 0.4112 | 209.60 |
| +Verapamil (5 μM) | 0.0167 ± 0.0027 | 0.85 | 0.0876 ± 0.0015 | 4.47 ** |
| +SSJ20 (5 μM) | 0.0189 ± 0.0019 | 0.96 | 4.2660 ± 0.4016 | 217.65 |
| +SSJ25 (5 μM) | 0.0194 ± 0.0018 | 0.99 | 4.0939 ± 0.0083 | 208.87 |
| +SSJ26 (5 μM) | 0.0176 ± 0.0017 | 0.90 | 0.0941 ± 0.5238 | 4.80 ** |
| +SSJ27 (5 μM) | 0.0169 ± 0.0019 | 0.86 | 3.4125 ± 0.3317 | 174.11 * |
| +SSJ28 (5 μM) | 0.0202 ± 0.0012 | 1.03 | 4.2550 ± 0.3567 | 217.09 |
| +SSJ29 (5 μM) | 0.0195 ± 0.0018 | 1.00 | 3.1563 ± 0.4173 | 161.04 * |
| +SSJ30 (5 μM) | 0.0190 ± 0.0021 | 0.97 | 4.2027 ± 0.3513 | 214.42 |
| +SSJ31 (5 μM) | 0.0186 ± 0.0016 | 0.95 | 4.2814 ± 0.5122 | 218.44 |
| +SSJ32 (5 μM) | 0.0188 ± 0.0019 | 0.96 | 0.1312 ± 0.0101 | 6.69 ** |
| +SSJ33 (5 μM) | 0.0195 ± 0.0020 | 0.99 | 2.9758 ± 0.2593 | 151.83 * |
| +SSJ34 (5 μM) | 0.0218 ± 0.0019 | 1.11 | 4.2762 ± 0.4135 | 218.17 |
| +SSJ35 (5 μM) | 0.0231 ± 0.0023 | 1.18 | 4.3574 ± 0.5143 | 222.32 |
| +SSJ36 (5 μM) | 0.0169 ± 0.0018 | 0.86 | 3.4676 ± 0.4491 | 176.92 |
| +SSJ37 (5 μM) | 0.0167 ± 0.0015 | 0.85 | 3.9948 ± 0.4798 | 203.82 |
3.2. Effect of SSJ26 and SSJ32 in P-GP Overexpressing Cancer and ABCB1-Transfected Cells
| Treatment | SW620 | SW620/Ad300 | ||
|---|---|---|---|---|
| IC50 ± SD a (μM) | FR b | IC50 ± SD a (μM) | FR b | |
| Doxorubicin | 0.0201 ± 0.0023 | (1.00) | 4.3221 ± 0.4513 | (215.16) |
| +SSJ26 (1.25 μM) | 0.0199 ± 0.0018 | (0.99) | 0.5314 ± 0.05134 | (26.44) * |
| +SSJ26 (2.5 μM) | 0.0185 ± 0.0017 | (0.92) | 0.3212 ± 0.09021 | (15.98) * |
| +SSJ26 (5 μM) | 0.0181 ± 0.0021 | (0.90) | 0.1017 ± 0.01022 | (5.06) ** |
| +SSJ32 (1.25 μM) | 0.0210 ± 0.0025 | (1.05) | 0.6354 ± 0.0724 | (31.61) * |
| +SSJ32 (2.5 μM) | 0.0199 ± 0.0021 | (0.99) | 0.3317 ± 0.0413 | (16.50) * |
| +SSJ32 (5 μM) | 0.0189 ± 0.0017 | (0.94) | 0.1528 ± 0.0132 | (7.60) ** |
| +Verapamil (5 μM) | 0.0179 ± 0.0015 | (0.89) | 0.0923 ± 0.0058 | (4.59) ** |
| Paclitaxel | 0.0072 ± 0.0010 | (1.00) | 2.7402 ± 0.2813 | (380.58) |
| +SSJ26 (1.25 μM) | 0.0063 ± 0.0008 | (0.88) | 0.2046 ± 0.0194 | (28.42) * |
| +SSJ26 (2.5 μM) | 0.0063 ± 0.0009 | (0.92) | 0.1081 ± 0.0098 | (15.01) * |
| +SSJ26 (5 μM) | 0.0057 ± 0.0006 | (0.79) * | 0.0737 ± 0.0084 | (10.23) ** |
| +SSJ32 (1.25 μM) | 0.0068 ± 0.0005 | (0.94) | 0.2977 ± 0.0313 | (41.35) * |
| +SSJ32 (2.5 μM) | 0.0064 ± 0.0006 | (0.89) | 0.1110 ± 0.0092 | (15.41) * |
| +SSJ32 (5 μM) | 0.0061 ± 0.0005 | (0.85) | 0.0872 ± 0.0095 | (12.12) ** |
| +Verapamil (5 μM) | 0.0062 ± 0.0007 | (0.86) | 0.0658 ± 0.0068 | (9.14) ** |
| Cisplatin | 1.8203 ± 0.1744 | (1.00) | 2.3217 ± 0.3353 | (1.28) |
| +SSJ26 (5 μM) | 1.8556 ± 0.2919 | (1.02) | 2.1842 ± 0.3148 | (1.20) |
| +SSJ32 (5 μM) | 1.7453 ± 0.2434 | (0.96) | 2.1772 ± 0.0210 | (1.19) |
| +Verapamil (5 μM) | 1.8012 ± 0.2017 | (0.99) | 2.0313 ± 0.0251 | (1.12) |
| Treatment | HEK293/pcDNA3.1 | HEK/ABCB1 | ||
|---|---|---|---|---|
| IC50 ± SD a (μM) | FR b | IC50 ± SD a (μM) | FR b | |
| Paclitaxel | 0.0224 ± 0.0030 | (1.00) | 0.9912 ± 0.1504 | (44.25) |
| +SSJ26 (1.25 μM) | 0.0279 ± 0.0047 | (1.25) | 0.1980 ± 0.0586 | (8.84) ** |
| +SSJ26 (5 μM) | 0.0242 ± 0.0019 | (1.08) | 0.0389 ± 0.0040 | (1.74) ** |
| +SSJ32 (1.25 μM) | 0.0254 ± 0.0025 | (1.13) | 0.2824 ± 0.016 | (12.61) * |
| +SSJ32 (5 μM) | 0.0162 ± 0.026 | (0.73) | 0.0582 ± 0.051 | (2.59) ** |
| +Verapamil (5 μM) | 0.0179 ± 0.0043 | (0.80) | 0.0415 ± 0.0055 | (1.85) ** |

3.3. Effect of SSJ26 and SSJ32 on Transfected HEK/ABCC1 and HEK/ABCG2 Cells
| Treatment | HEK293/pcDNA3.1 | HEK293/ABCC1 | ||
|---|---|---|---|---|
| IC50 ± SD a (μM) | FR b | IC50 ± SD a (μM) | FR b | |
| Vincristine | 0.0208 ± 0.0021 | (1.00) | 0.4731 ± 0.0282 | (22.75) |
| +SSJ26 (5 μM) | 0.0193 ± 0.0019 | (0.93) | 0.4693 ± 0.0184 | (22.56) |
| +SSJ32 (5 μM) | 0.0235 ± 0.0027 | (1.13) | 0.4897 ± 0.0639 | (23.54) |
| +PAK-104P c (5 μM) | 0.0189 ± 0.0023 | (0.91) | 0.0778 ± 0.0092 | (3.74) ** |
| Treatment | HEK293/pcDNA3.1 | HEK293/ABCG2 | ||
| IC50 ± SD a (μM) | FR b | IC50 ± SD a (μM) | FR b | |
| Mitoxantrone | 0.0309 ± 0.0023 | (1.0) | 0.4259 ± 0.0203 | (13.8) |
| +SSJ26 (5 μM) | 0.0294 ± 0.0019 | (0.95) | 0.3729 ± 0.0553 | (12.0) |
| +SSJ32 (5 μM) | 0.0333 ± 0.0028 | (1.08) | 0.3654 ± 0.0675 | (11.8) |
| +FTC d (5 μM) | 0.0273 ± 0.0032 | (0.88) | 0.0381 ± 0.0435 | (1.2) ** |
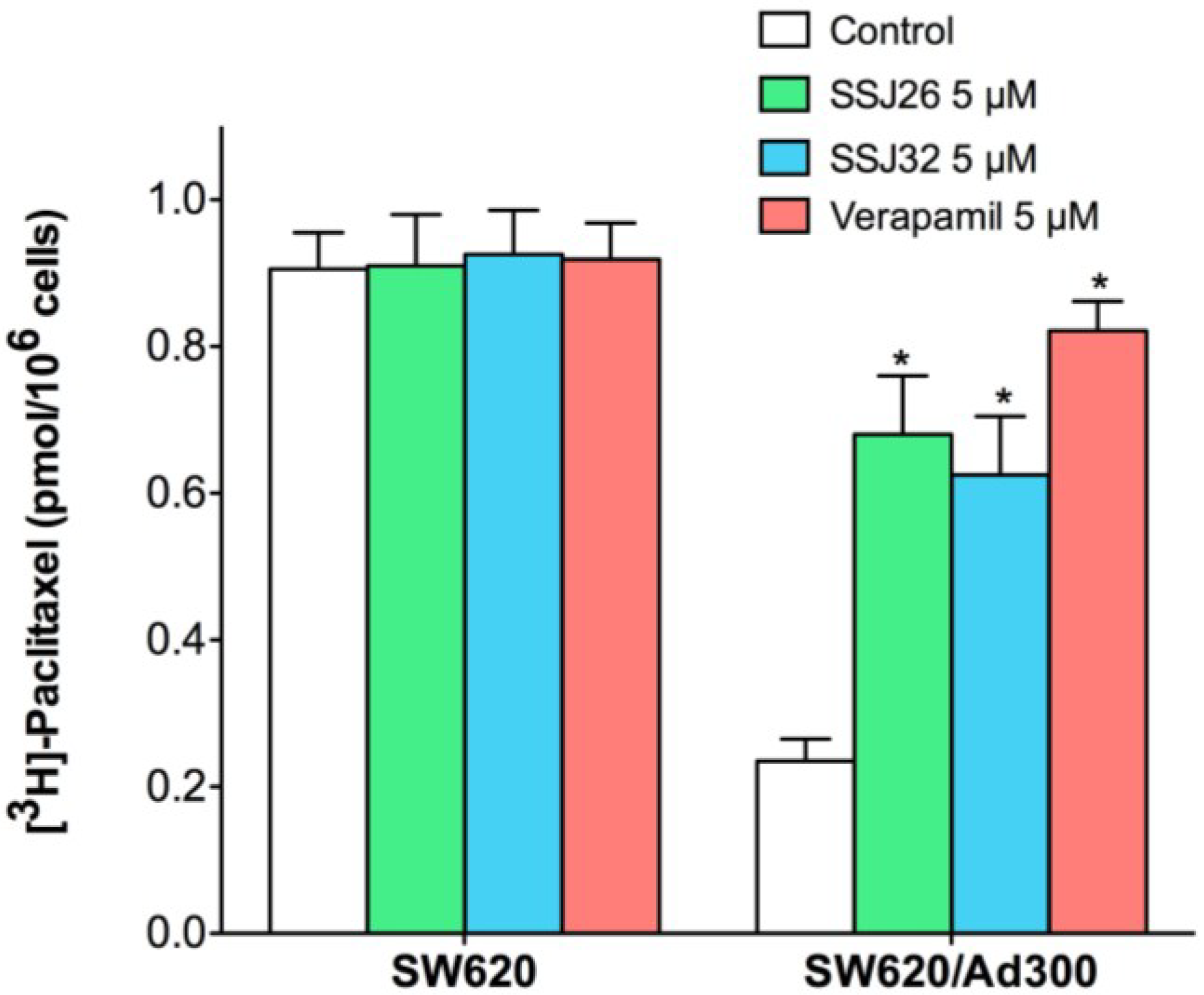
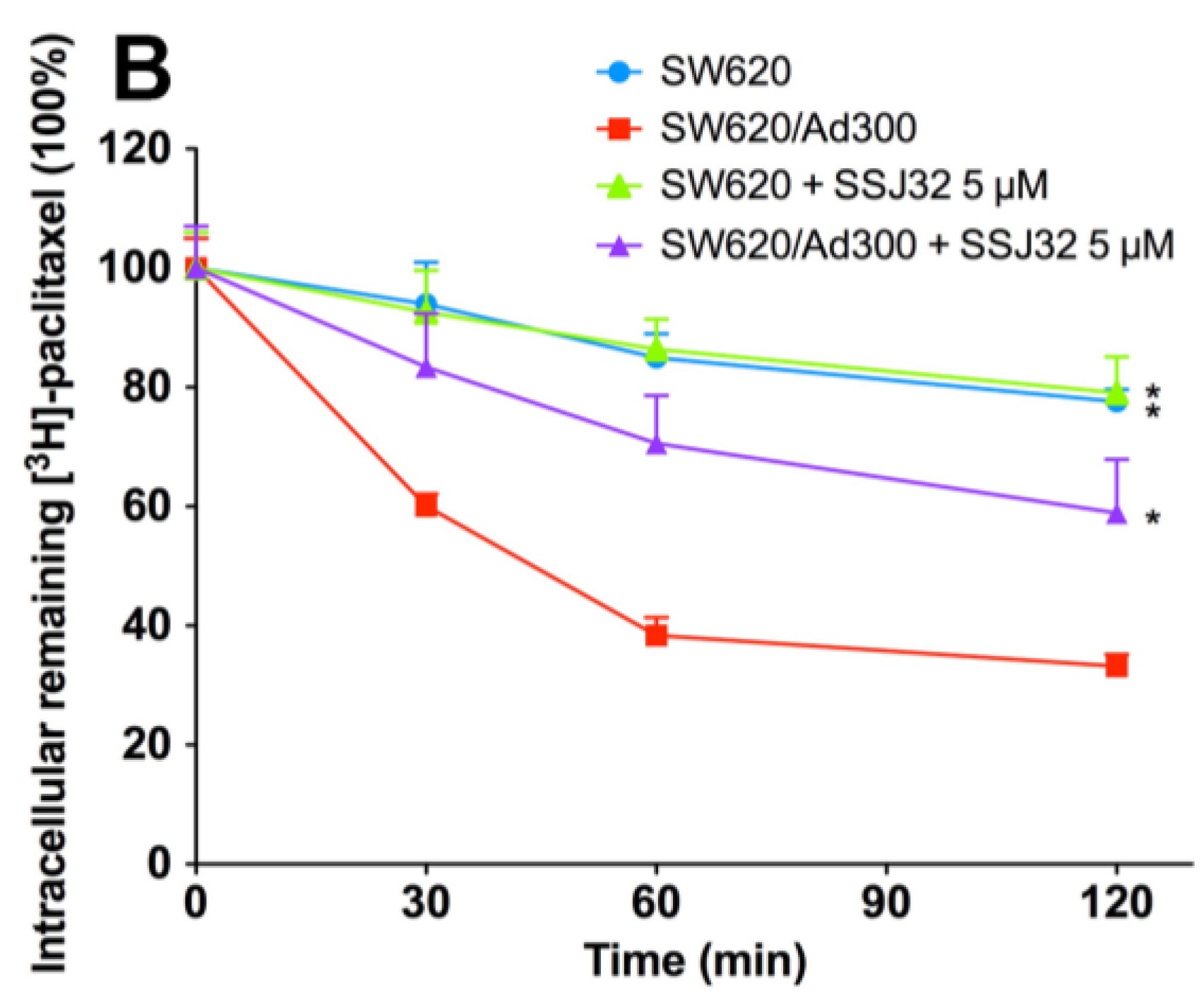
3.4. SSJ26 and SSJ32 Increased Intracellular Accumulation of [3H]-Paclitaxel by Inhibiting Its Efflux


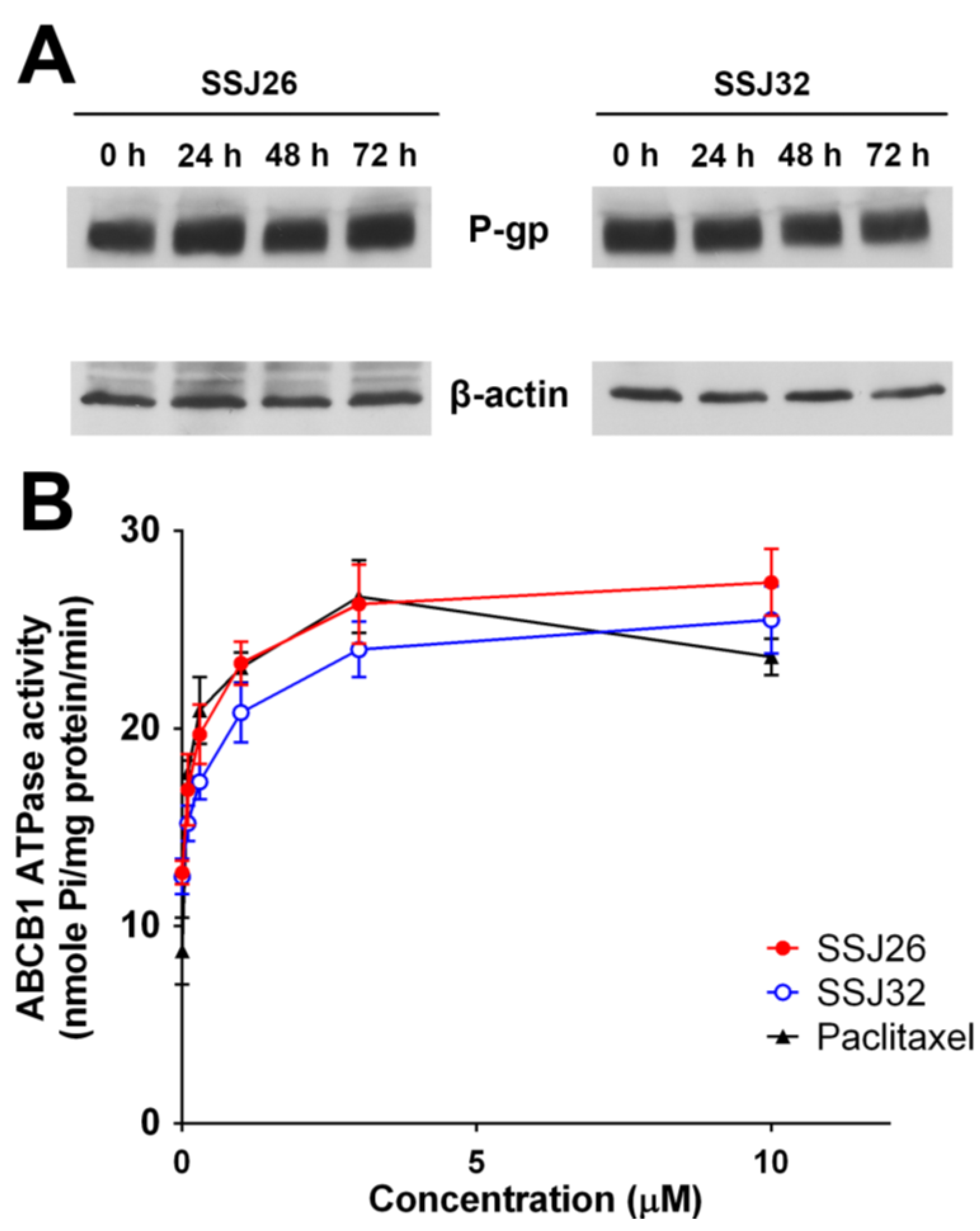
3.5. SSJ26 and SSJ32 Stimulated ATPase Activities but Did not Alter the Expression Level of P-GP

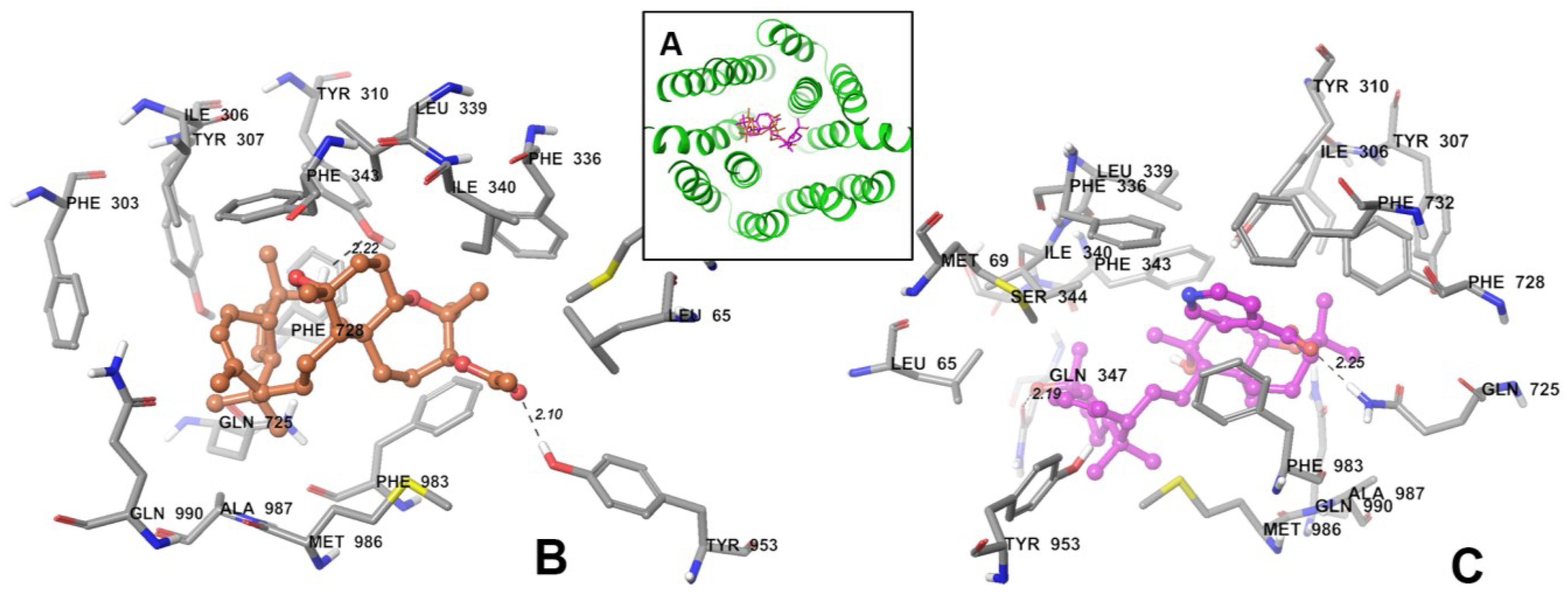
3.6. Binding Mode of SSJ26 and SSJ32 with Homology Model of P-GP

4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R., Jr.; Chen, Z.S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updat.: Rev. Comment. Antimicrob. Anticancer Chemother. 2014, 18, 1–17. [Google Scholar] [CrossRef]
- Anreddy, N.; Gupta, P.; Kathawala, R.J.; Patel, A.; Wurpel, J.N.; Chen, Z.S. Tyrosine kinase inhibitors as reversal agents for ABC transporter mediated drug resistance. Molecules 2014, 19, 13848–13877. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; Chang, G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.; Coley, H.M. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003, 10, 159–165. [Google Scholar] [PubMed]
- Tiwari, A.K.; Sodani, K.; Dai, C.L.; Ashby, C.R., Jr.; Chen, Z.S. Revisiting the ABCs of multidrug resistance in cancer chemotherapy. Curr. Pharm. Biotechnol. 2011, 12, 570–594. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Zhang, Y.K.; Kathawala, R.J.; Chen, Z.S. Repositioning of Tyrosine Kinase Inhibitors as Antagonists of ATP-Binding Cassette Transporters in Anticancer Drug Resistance. Cancers 2014, 6, 1925–1952. [Google Scholar] [CrossRef] [PubMed]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981, 41, 1967–1972. [Google Scholar] [PubMed]
- Twentyman, P.R.; Fox, N.E.; White, D.J. Cyclosporin A and its analogues as modifiers of adriamycin and vincristine resistance in a multi-drug resistant human lung cancer cell line. Br. J. Cancer 1987, 56, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.; Higgins, C.F. Interaction of tamoxifen with the multidrug resistance P-glycoprotein. Br. J. Cancer 1995, 71, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.R.; George, S.L.; Dodge, R.K.; O’Loughlin, K.L.; Minderman, H.; Caligiuri, M.A.; Anastasi, J.; Powell, B.L.; Kolitz, J.E.; Schiffer, C.A.; et al. Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia: Cancer and Leukemia Group B Study 9720. Blood 2002, 100, 1224–1232. [Google Scholar] [PubMed]
- Benet, L.Z.; Cummins, C.L.; Wu, C.Y. Unmasking the dynamic interplay between efflux transporters and metabolic enzymes. Int. J. Pharm. 2004, 277, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Products 2004, 67, 1216–1238. [Google Scholar] [CrossRef]
- Huang, X.C.; Xiao, X.; Zhang, Y.K.; Talele, T.T.; Salim, A.A.; Chen, Z.S.; Capon, R.J. Lamellarin O, a pyrrole alkaloid from an Australian marine sponge, Ianthella sp., reverses BCRP mediated drug resistance in cancer cells. Mar. Drugs 2014, 12, 3818–3837. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; El Sayed, K.; Chen, Z.S.; Guo, H. Current status on marine products with reversal effect on cancer multidrug resistance. Mar. Drugs 2012, 10, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Funel-Le Bon, C.; Berrue, F.; Thomas, O.P.; Reyes, F.; Amade, P. Sodwanone S, a triterpene from the marine sponge Axinella weltneri. J. Nat. Products 2005, 68, 1284–1287. [Google Scholar]
- Rudi, A.; Aknin, M.; Gaydou, E.M.; Kashman, Y. Sodwanones K, L, and M; new triterpenes from the marine sponge Axinella weltneri. J. Nat. Products 1997, 60, 700–703. [Google Scholar] [CrossRef]
- Foudah, A.I.; Jain, S.; Busnena, B.A.; El Sayed, K.A. Optimization of marine triterpene sipholenols as inhibitors of breast cancer migration and invasion. ChemMedChem 2013, 8, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Foudah, A.I.; Sallam, A.A.; Akl, M.R.; El Sayed, K.A. Optimization, pharmacophore modeling and 3D-QSAR studies of sipholanes as breast cancer migration and proliferation inhibitors. Eur. J. Med. Chem. 2014, 73, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Tiwari, A.K.; Chufan, E.E.; Sodani, K.; Anreddy, N.; Singh, S.; Ambudkar, S.V.; Stephani, R.; Chen, Z.S. PD173074, a selective FGFR inhibitor, reverses ABCB1-mediated drug resistance in cancer cells. Cancer Chemother. Pharmacol. 2013, 72, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.J.; Zhang, Y.K.; Wang, D.S.; Kathawala, R.J.; Patel, A.; Talele, T.T.; Chen, Z.S.; Fu, L.W. AST1306, a potent EGFR inhibitor, antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance. Cancer Lett. 2014, 350, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.Q.; Zhang, G.N.; Wang, Y.J.; Zhang, Y.K.; Sodani, K.; Talele, T.T.; Ashby, C.R., Jr.; Chen, Z.S. β-Elemene, a compound derived from Rhizoma zedoariae, reverses multidrug resistance mediated by the ABCB1 transporter. Oncol. Rep. 2014, 31, 858–866. [Google Scholar] [PubMed]
- Zhang, H.; Zhang, Y.K.; Wang, Y.J.; Kathawala, R.J.; Patel, A.; Zhu, H.; Sodani, K.; Talele, T.T.; Ambudkar, S.V.; Chen, Z.S.; et al. WHI-P154 enhances the chemotherapeutic effect of anticancer agents in ABCG2-overexpressing cells. Cancer Sci. 2014, 105, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Sodani, K.; Patel, A.; Anreddy, N.; Singh, S.; Yang, D.H.; Kathawala, R.J.; Kumar, P.; Talele, T.T.; Chen, Z.S. Telatinib reverses chemotherapeutic multidrug resistance mediated by ABCG2 efflux transporter in vitro and in vivo. Biochem. Pharmacol. 2014, 89, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wang, R.; Wang, J.; Hua, K.; Wang, Y.; Liu, X.; Yao, S. ALT1, a Snf2 family chromatin remodeling ATPase, negatively regulates alkaline tolerance through enhanced defense against oxidative stress in rice. PLoS ONE 2014, 9, e112515. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Kathawala, R.J.; Zhang, Y.K.; Patel, A.; Kumar, P.; Shukla, S.; Fung, K.L.; Ambudkar, S.V.; Talele, T.T.; Chen, Z.S. Motesanib (AMG706), a potent multikinase inhibitor, antagonizes multidrug resistance by inhibiting the efflux activity of the ABCB1. Biochem. Pharmacol. 2014, 90, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaimes, K.; Aller, S. Refined structures of mouse P-Glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; Jain, S.; Wu, C.P.; Khanfar, M.A.; Kuang, Y.; Dai, C.L.; Shi, Z.; Chen, X.; Fu, L.; Ambudkar, S.V.; et al. Marine sponge-derived sipholane triterpenoids reverse P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Biochem. Pharmacol. 2010, 80, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Sodani, K.; Tiwari, A.K.; Singh, S.; Patel, A.; Xiao, Z.J.; Chen, J.J.; Sun, Y.L.; Talele, T.T.; Chen, Z.S. GW583340 and GW2974, human EGFR and HER-2 inhibitors, reverse ABCG2- and ABCB1-mediated drug resistance. Biochem. Pharmacol. 2012, 83, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhang, Y.-K.; Wang, Y.-J.; Vispute, S.G.; Jain, S.; Chen, Y.; Li, J.; Youssef, D.T.A.; Sayed, K.A.E.; Chen, Z.-S. Esters of the Marine-Derived Triterpene Sipholenol A Reverse P-GP-Mediated Drug Resistance. Mar. Drugs 2015, 13, 2267-2286. https://doi.org/10.3390/md13042267
Zhang Y, Zhang Y-K, Wang Y-J, Vispute SG, Jain S, Chen Y, Li J, Youssef DTA, Sayed KAE, Chen Z-S. Esters of the Marine-Derived Triterpene Sipholenol A Reverse P-GP-Mediated Drug Resistance. Marine Drugs. 2015; 13(4):2267-2286. https://doi.org/10.3390/md13042267
Chicago/Turabian StyleZhang, Yongchao, Yun-Kai Zhang, Yi-Jun Wang, Saurabh G. Vispute, Sandeep Jain, Yangmin Chen, Jessalyn Li, Diaa T. A. Youssef, Khalid A. El Sayed, and Zhe-Sheng Chen. 2015. "Esters of the Marine-Derived Triterpene Sipholenol A Reverse P-GP-Mediated Drug Resistance" Marine Drugs 13, no. 4: 2267-2286. https://doi.org/10.3390/md13042267
APA StyleZhang, Y., Zhang, Y. -K., Wang, Y. -J., Vispute, S. G., Jain, S., Chen, Y., Li, J., Youssef, D. T. A., Sayed, K. A. E., & Chen, Z. -S. (2015). Esters of the Marine-Derived Triterpene Sipholenol A Reverse P-GP-Mediated Drug Resistance. Marine Drugs, 13(4), 2267-2286. https://doi.org/10.3390/md13042267