3. Experimental Section
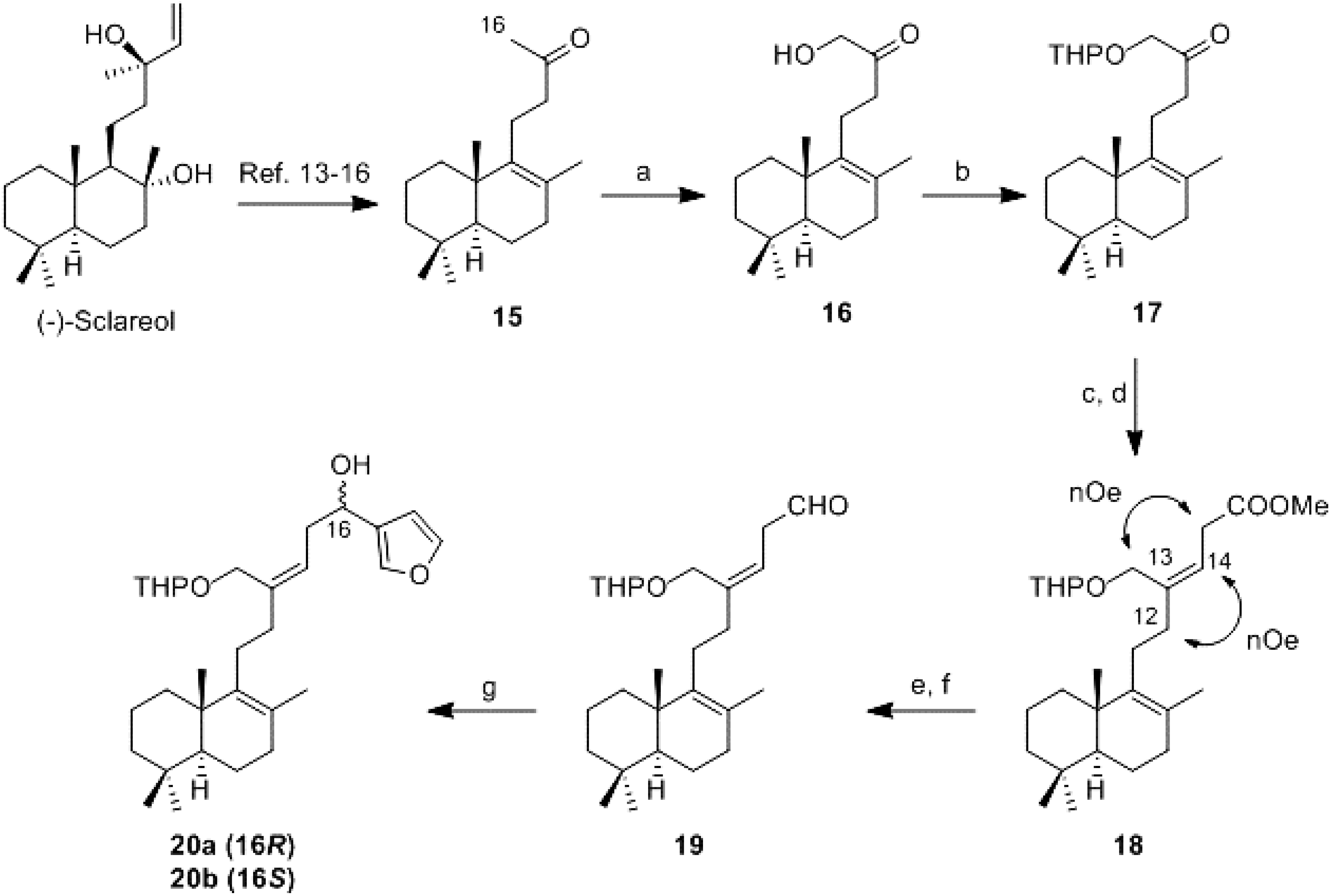
16-Hydroxy-14,15-dinor-labd-8-en-13-one (16): To a stirred solution of 15 (60 mg, 0.23 mmol) in MeOH (1.1 mL) was added slowly a solution of KOH (76 mg, 1.15 mmol) in MeOH (1.75 mL) and the mixture was reacted at 0 °C for 10 min. Afterwards, (diacetoxyiodo)benzene (DIB) (146 mg, 0.46 mmol) was added, and the mixture was stirred at 0 °C following the reaction evolution by TLC. When the reaction had finished, a 5% aqueous solution of H2SO4 (1.5 mL) was added and the mixture was reacted at 0 °C for 90 min. It was quenched with water and the product was extracted with DCM. The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo. The resulting crude residue was purified by flash CC (hexane-AcOEt, 98:2) to obtain 16 (33 mg, 50%).
= +81.9 (c 0.64, CHCl3); IR υ 3443 (OH), 2936, 1721 (C=O), 1441, 1375, 1024; 1H-NMR (400 MHz, CDCl3) δ 4.24 (2H, s, H-16), 2.50–2.30 (4H, m, H-11, H-12), 2.00–1.00 (11H, m), 1.57 (3H, s, Me-17), 0.94 (3H, s, Me-20), 0.88 (3H, s, Me-18), 0.82 (3H, s, Me-19); 13C-NMR (100 MHz, CDCl3) δ 209.6 (C), 138.7 (C), 127.3 (C), 67.9 (CH2), 51.9 (CH), 41.7 (CH2), 39.3 (CH2), 39.0 (C), 37.0 (CH2), 33.6 (CH2), 33.3 (C, CH3), 21.6 (CH3), 21.4 (CH2), 19.9 (CH3), 19.4 (CH3), 18.9 (CH2-2).
16-(2-Tetrahydropyranyloxy)-14,15-dinor-labd-8-en-13-one (17): To a stirred solution of 16 (152 mg, 0.54 mmol) in dry benzene (3.6 mL) was added p-toluenesulfonic acid (3 mg, 0.016 mmol) and dihydropyran (DHP) (0.15 mL, 1.62 mmol). The evolution of reaction was controlled by TLC. When the reaction had finished a 10% aqueous solution of Na2CO3 (3 mL) was added and it was reacted for 30 min. It was quenched with water and the product was extract with AcOEt. The combined organic layers were washed with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo to obtain 17 (196 mg, 100%).
= +26.0 (c 2.4, CHCl3); IR υ 2941, 1717 (C=O), 1665 (C=C), 1456, 1126, 1036; 1H-NMR (400 MHz, CDCl3) δ 4.94 (1H, dd, J = 2.8 and 4.8 Hz, H-2′ major.), 4.63 (1H, t, J = 3.7 Hz, H-2′ minor.), 4.23 (1H, d, J = 17 Hz, HA-16), 4.09 (1H, d, J = 17 Hz, HB-16), 3.90–3.40 (2H, m, H-6′), 2.60–2.50 (2H, m, H-12), 2.40–2.10 (2H, m, H-11), 2.00–1.00 (17H, m), 1.53 (3H, s, Me-17), 0.93 (3H, s, Me-20), 0.87 (3H, s, Me-18), 0.82 (3H, s, Me-19); 13C-NMR (100 MHz, CDCl3) δ 208.8 (C), 139.2 (C), 126.7 (C), 98.9/94.6 (CH), 71.9 (CH2), 62.9/62.4 (CH2), 51.9 (CH), 41.7 (CH2), 39.9 (CH2), 39.0 (C), 36.9 (CH2), 33.6 (CH2), 33.2 (C, CH3), 30.6/30.2 (CH2), 25.4/25.2 (CH2), 21.6 (CH3), 21.3 (CH2), 19.9 (CH3), 19.7/19.2 (CH2), 19.4 (CH3), 18.9 (CH2-2); HRMS (ESI) m/z calcd for C23H38O3Na (M + Na)+ 385.2713, found 385.2722.
Methyl 21-(2-tetrahydropyranyloxy)-17,18,19,20-tetranor-luffara-8,13Z-dien-16-oate (18): To a suspension of (2-carboxyethyl)triphenylphosphonium bromide (270 mg, 0.65 mmol) in dry THF (3.2 mL) and dry DMSO (0.8 mL) at −5 °C under argon atmosphere, n-BuLi (1.6 M in hexane; 0.8 mL, 1.3 mmol) was added slowly and the reaction was stirred for 10 min. A solution of 17 (47 mg, 0.13 mmol) in THF/DMSO 4:1 (2.5 mL) was added via cannula dropwise and the reaction was stirred for 90 min. It was allowed to warm to room temperature, quenched with saturated aqueous solution of NH4Cl and extracted with AcOEt. The combined organic layers were washed with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The obtained acid was directly esterified: the resulting crude residue was dissolved in C6H6/MeOH 1:1 (2.4 mL) and cooled at 0 °C. Under argon atmosphere TMSCHN2 (2.0 M in hexane; 0.3 mL, 0.6 mmol) was added. After 10 min, the solvent was removed under reduced pressure and the resulting crude residue was purified by CC (hexane-AcOEt, 97:3) to obtain 18 (30 mg, 54%).
= +41.4 (c 0.8, CHCl3); IR υ 2941, 2868, 1744 (C=O), 1200, 1132, 1024; 1H-NMR (400 MHz, CDCl3) δ 5.58 (1H, t, J = 7.1 Hz, H-14), 4.58 (1H, t, J = 3.0 Hz, H-2′), 4.21 (1H, dd, J = 11.9 Hz, HA-21), 4.04 (1H, dd, J = 2.1 and 11.9 Hz, HB-21), 3.90–3.80 (1H, m, H-6′), 3.68 (3H, s, COOMe), 3.50–3.45 (1H, m, H-6′), 3.17 (2H, d, J = 7.1 Hz, H-15), 2.20–2.05 (4H, m, H-12, H-11), 1.95–1.10 (17H, m), 1.57 (3H, s, Me-22), 0.93 (3H, s Me-23), 0.88 (3H, s, Me-25), 0.82 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 172.5 (C), 140.2 (C), 136.8 (C), 126.0 (C), 119.5 (CH), 97.6/94.6 (CH), 64.3 (CH2), 62.9/62.1 (CH2), 51.9 (CH), 51.7 (CH3), 41.8 (CH2), 39.0 (C), 36.9 (CH2), 36.0 (CH2), 33.6 (CH2), 33.3 (C, CH3), 33.1 (CH2), 30.6/30.5 (CH2), 27.1 (CH2), 25.4 (CH2), 21.7 (CH3), 20.0 (CH3), 19.7 (CH2), 19.5 (CH3), 19.0 (CH2 - 2); HRMS (ESI) m/z calcd for C27H44O4Na (M + Na)+ 455.3132, found 455.3116.
21-(2-Tetrahydropyranyloxy)-17,18,19,20-tetranor-luffara-8,13Z-dien-16-al (19): To a solution of 18 (31 mg, 0.072 mmol) in Et2O (5.3 mL) at 0 °C was added LiAlH4 (27 mg, 0.72 mmol). The reaction was stirred at rt for 15 min and then quenched with wet AcOEt, dried (Na2SO4), filtered through a short pad of Celite and concentrated in vacuo. The resulting alcohol (80 mg, 0.196 mmol) was solved in DCM (11.8 mL) and it was added DMP (103 mg, 0.25 mmol). The reaction was stirred at rt for 30 min. It was diluted with AcOEt and washed with 10% NaHCO3/10% Na2S2O3 1:1, dried (Na2SO4), filtered, and concentrated in vacuo to obtain 19 (79 mg, 100% from 18). IR υ 2940, 2725, 1726 (CHO), 1684 (C=C), 1456, 1375, 1119, 1024; 1H-NMR (400 MHz, CDCl3) δ 9.67 (1H, s, H-16), 5.57 (1H, t, J = 7.4 Hz, H-14), 4.60–4.57 (1H, m, H-2′), 4.23 (1H, d, J = 11.8 Hz, HA-21), 4.03 (1H, d, J = 11.8 Hz, HB-21), 3.89–3.49 (2H, m, H-6′), 3.27 (2H, d, J = 7,5 Hz, H-15), 2.13–2.05 (4H, m, H-11, H-12), 1.95–1.10 (17H, m, H-7), 1.58 (3H, s, Me-22), 0.94 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.83 (3H, s, Me-24).
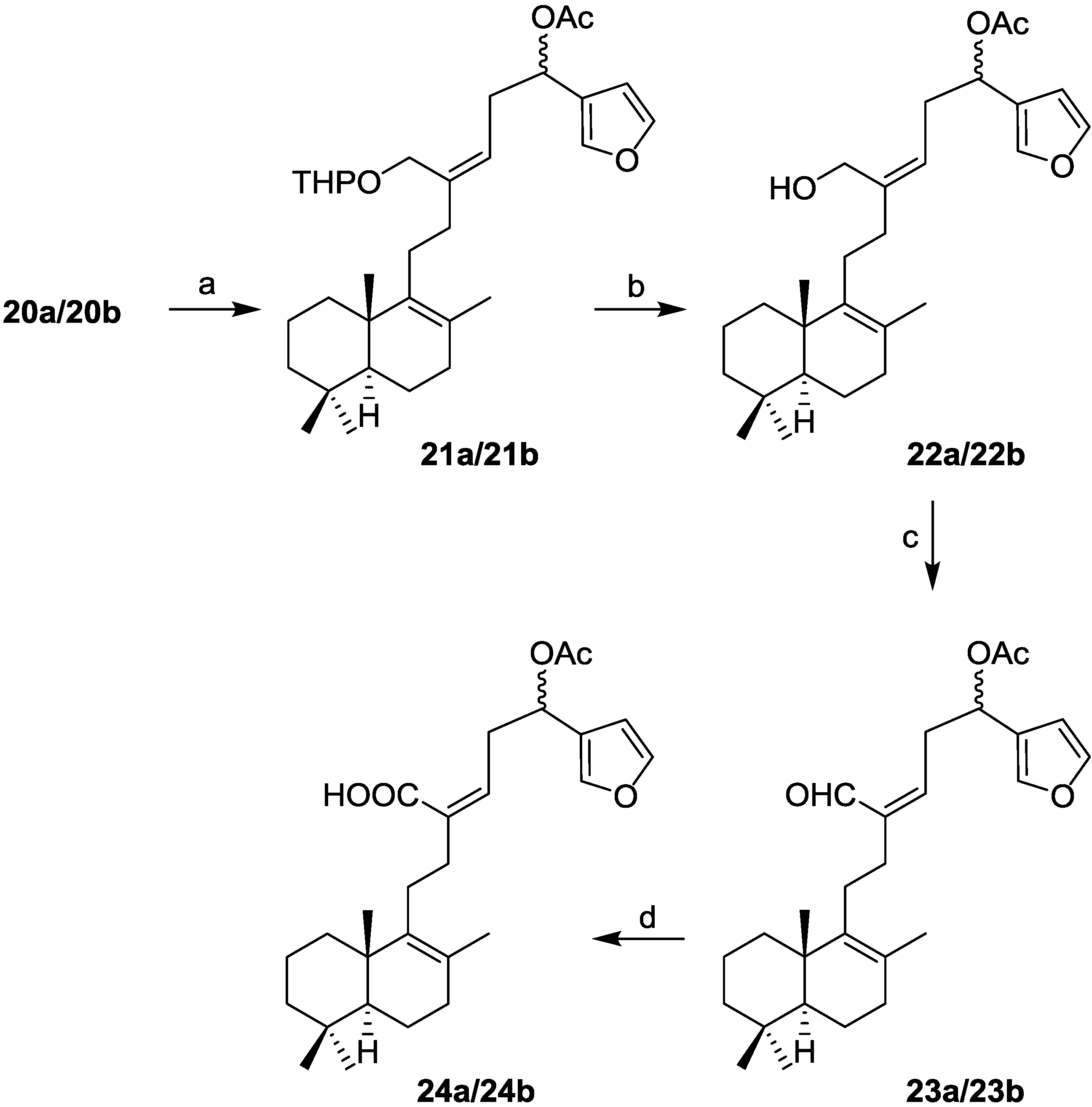
21-(2-Tetrahydropyranyloxy)-19,20-epoxy-luffara-8,13Z,17(20),18-tetraen-16(R,S)-ol (20a/20b): To a solution of 3-bromofuran (0.13 mL, 1.47 mmol) in Et2O at −78 °C under argon atmosphere was added dropwise n-BuLi (1.6 M in hexane; 0.92 mL, 1.47 mmol) and the solution was stirred for 10 min. After that, a solution of 19 (59 mg, 0.147 mmol) in Et2O (1.6 mL) was added dropwise via cannula and the mixture was stirred for 30 min. It was allowed to warm to room temperature, quenched with a saturated aqueous solution of NH4Cl and extracted with AcOEt. The combined organic layers were washed with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The resulting crude residue was purified by flash CC (hexane-AcOEt, 9:1) to obtain a mixture of 20a/20b (27 mg, 41%).
= +47.4 (c 0.22, CHCl3); IR υ 3249 (OH), 2940, 1440, 1202, 1024; 1H-NMR (400 MHz, CDCl3) δ 7.37 (2H, s, H-19, H-20), 6.38 (1H, s, H-18), 5.51–5.43 (1H, m, H-14), 4.71–4.64 (2H, m, H-2′, H-16), 4.17–3.90 (2H, m, H-21), 3.88–3.52 (2H, m, H-6′), 2.60–2.40 (2H, m, H-15), 2.20–2.10 (4H, m, H-11, H-12), 1.85–1.05 (17H, m), 1.58 (3H, s, Me-22), 0.94 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.83 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 143.1 (CH), 140.6 (C), 140.2 (C), 138.9/138.8 (CH), 129.2 (C), 126.0 (C), 124.7 (CH), 108.6 (CH), 97.9/97.2 (CH), 66.2 (CH), 64.6/64.2 (CH2), 61.9/61.7 (CH2), 51.9 (CH), 41.9 (CH2), 39.0 (C), 37.0 (CH2), 36.8 (CH2), 36.6 (CH2), 33.6 (CH2), 33.3 (C, CH3), 30.4/30.3 (CH2), 27.3 (CH2), 25.4 (CH2), 21.7 (CH3), 20.1 (CH3), 19.7 (CH2), 19.5 (CH3), 19.0 (CH2 - 2); HRMS (ESI) m/z calcd for C30H46O4Na (M + Na)+ 493.3288, found 493.3303.
16(R,S)-Acetoxy-21-(2-tetrahydropyranyloxy)-19,20-epoxi-luffara-8,13Z,17(20),18-tetraene (21a/21b): To a solution of 20a/20b (30 mg, 0.064 mmol) in pyridine (1.5 mL) was added acetic anhydride (1.5 mL) and the reaction was stirred at rt in anhydrous conditions for 24 h. It was quenched with ice and extracted with AcOEt. The combined organic layers were washed with 2 M aqueous solution of HCl, 10% aqueous solution of NaHCO3 and water until neutral pH was reached, dried (Na2SO4), filtered, and concentrated in vacuo to obtain 21a/21b (32 mg, 99%).
= +19.6 (c 0.08, CHCl3); IR υ 2940, 1742 (C=O), 1371, 1236, 1024; 1H-NMR (400 MHz, CDCl3) δ 7.41 (1H, s, H-20), 7.37 (1H, s, H-19), 6.39 (1H, bs, H-18), 5.80–5.75 (1H, m, H-16), 5.31 (1H, t, J = 7.3 Hz, H-14), 4.60–4.50 (1H, m, H-2′), 4.25–3.95 (2H, m, H-21), 3.95–3.45 (2H, m, H-6′), 2.80–2.50 (2H, m, H-15), 2.20–2.10 (4H, m, H-11, H-12), 2.04 (3H, s, MeCOO), 2.00–1.00 (17H, m), 1.57 (3H, s, Me-22), 0.93 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.82 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 170.3 (C), 143.1 (CH), 140.3 (C, CH), 140.2 (C), 126.0 (C), 123.5 (C), 122.5 (CH), 109.0 (CH), 97.8/97.6 (CH), 68.2 (CH), 64.2 (CH2), 62.1/61.9 (CH2), 51.9 (CH), 41.8 (CH2), 39.0 (C), 36.9 (CH2), 36.1 (CH2), 33.6 (CH2), 33.3 (C, CH3), 33.1 (CH2), 30.6 (CH2), 27.4 (CH2), 25.4 (CH2), 21.7 (CH3), 21.2 (CH3), 20.1 (CH3), 19.5 (CH3), 19.3 (CH2), 19.0 (CH2 - 2); HRMS (ESI) m/z calcd for C32H48O5Na (M + Na)+ 535.3394, found: 535.3381.
16(R,S)-Acetoxy-19,20-epoxy-luffara-8,13Z,17(20),18-tetraen-21-ol (22a/22b): To a solution of 21a/21b (65 mg, 0.13 mmol) in MeOH (12.2 mL) was added p-toluenesulfonic acid (8 mg, 0.04 mmol) and the reaction was stirred at rt for 4 h. It was added water and extracted with AcOEt. The combined organic layers were washed with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo to afford 22a/22b (56 mg, 100%).
= +8.9 (c 0.17, CHCl3); IR υ 3468 (OH), 3134, 2928, 1739 (C=O), 1371, 1238, 1024; 1H-NMR (400 MHz, CDCl3) δ 7.41 (1H, s, H-20), 7.37 (1H, s, H-19), 6.39 (1H, bs, H-18), 5.80–5.75 (1H, m, H-16), 5.31 (1H, t, J = 7.3 Hz, H-14), 4.12 (1H, d, J = 14.0 Hz, HA-21), 4.11 (1H, d, J = 14.0 Hz, HB-21), 2.80–2.50 (2H, m, H-15), 2.20–2.10 (4H, m, H-11, H-12), 2.04 (3H, s, MeCOO), 2.00–1.00 (11H, m), 1.57 (3H, s, Me-22), 0.93 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.82 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 170.3 (C), 143.3 (CH), 142.9 (C), 140.3 (C, CH), 126.0 (C), 124.1 (C), 121.8 (CH), 108.8 (CH), 68.3 (CH), 60.1 (CH2), 51.8 (CH), 41.7 (CH2), 38.9 (C), 36.9 (CH2), 36.3 (CH2), 33.6 (CH2), 33.3 (C, CH3), 33.1 (CH2), 27.3 (CH2), 21.7 (CH3), 21.2 (CH3), 20.1 (CH3), 19.5 (CH3), 19.0 (CH2-2); HRMS (ESI) m/z calcd for C27H40O4Na (M + Na)+ 451.2819, found 451.2804.
16-(R,S)-Acetoxy-19,20-epoxy-luffara-8,13Z,17(20),18-tetraen-21-al (23a/23b): To a solution of 22a/22b (10 mg, 0.025 mmol) in DCM (1.4 mL) was added DMP (13 mg, 0.05 mmol). The reaction was stirred at rt for 30 min. It was added AcOEt and washed with 10% NaHCO3/10% Na2S2O3 1:1, dried (Na2SO4), filtered, and concentrated in vacuo to obtain 23a/23b (11 mg, 99%).
= +22.2 (c 0.1, CHCl3); IR υ 3136, 2929, 2868 (CHO), 2733, 1741 (C=O), 1678 (C=C), 1371, 1234, 1024; 1H-NMR (400 MHz, CDCl3) δ 10.07 (1H, s, H-21), 7.43 (1H, s, H-20), 7.41 (1H, s, H-19), 6.40 (1H, bs, H-18), 6.38 (1H, t, J = 8.4 Hz, H-14), 5.91 (1H, t, J = 6.6 Hz, H-16), 3.22–2.95 (2H, m, H-15), 2.20–2.00 (4H, m, H-11, H-12), 2.05 (3H, s, MeCOO), 2.00–1.00 (11H, m), 1.58 (3H, s, Me-22), 0.92 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.83 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 190.7 (C), 171.1 (C), 143.6 (CH), 143.4 (C), 141.3 (CH), 140.2 (CH), 139.8 (C), 126.7 (C), 123.8 (C), 108.6 (CH), 67.3 (CH), 51.9 (CH), 41.7 (CH2), 39.0 (C), 36.9 (CH2), 33.6 (CH2), 33.3 (C, CH3), 33.1 (CH2), 31.6 (CH2), 27.8 (CH2), 21.7 (CH3), 21.0 (CH3), 20.0 (CH3), 19.5 (CH3), 19.0 (CH2 - 2); HRMS (ESI) m/z calcd for C27H38O4Na (M + Na)+ 449.2662, found 449.2660.
16-(R,S)-Acetoxy-19,20-epoxy-luffara-8,13Z,17(20),18-tetraen-21-oic acid (24a/24b): To a solution of 23a/23b (8 mg, 0.019 mmol) in t-BuOH (0.25 mL) and 2-methyl-2-butene (51 μL), a solution of monobasic sodium phosphate (NaH2PO4, 10 mg) in water (0.1 mL) and 5% aqueous solution of NaClO2 (48 μL) were added. The reaction mixture was stirred at rt for 30 min. Then, water and 2 M aqueous solution of HCl were added. It was extracted with AcOEt and the combined organic layers were washed with water until neutral pH was reached, dried (Na2SO4), filtered, and concentrated in vacuo to afford 24a/24b (8 mg, 99%).
= +31.4 (c 0.5, CHCl3); IR υ 3500–2700 (COOH), 2924, 2855, 1744 (C=O), 1694 (C=O), 1645 (C=C), 1456, 1371, 1234, 1024; 1H-NMR (400 MHz, CDCl3) δ 7.43 (1H, s, H-20), 7.38 (1H, s, H-19), 6.40 (1H, bs, H-18), 5.98 (1H, t, J = 6.9 Hz, H-14), 5.89 (1H, t, J = 7.0 Hz, H-16), 3.15–3.00 (2H, m, H-15), 2.35–2.20 (4H, m, H-11, H-12), 2.06 (3H, s, MeCOO), 1.80–1.00 (11H, m), 1.56 (3H, s, Me-22), 0.92 (3H, s, Me-23), 0.87 (3H, s, Me-25), 0.82 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 171.8 (C), 170.1 (C), 143.1 (CH), 140.4 (CH), 139.3 (C), 138.3 (CH), 133.9 (C), 126.5 (C), 124.0 (C), 108.6 (CH), 67.8 (CH), 51.9 (CH), 41.8 (CH2), 39.0 (C), 36.9 (CH2), 35.1 (CH2), 34.5 (CH2), 33.6 (CH2), 33.3 (C, CH3), 27.8 (CH2), 21.7 (CH3), 21.1 (CH3), 20.1 (CH3), 19.5 (CH3), 19.0 (CH2-2); HRMS (ESI) m/z calcd for C27H38O5Na (M + Na)+ 465.2611, found 465.2604.
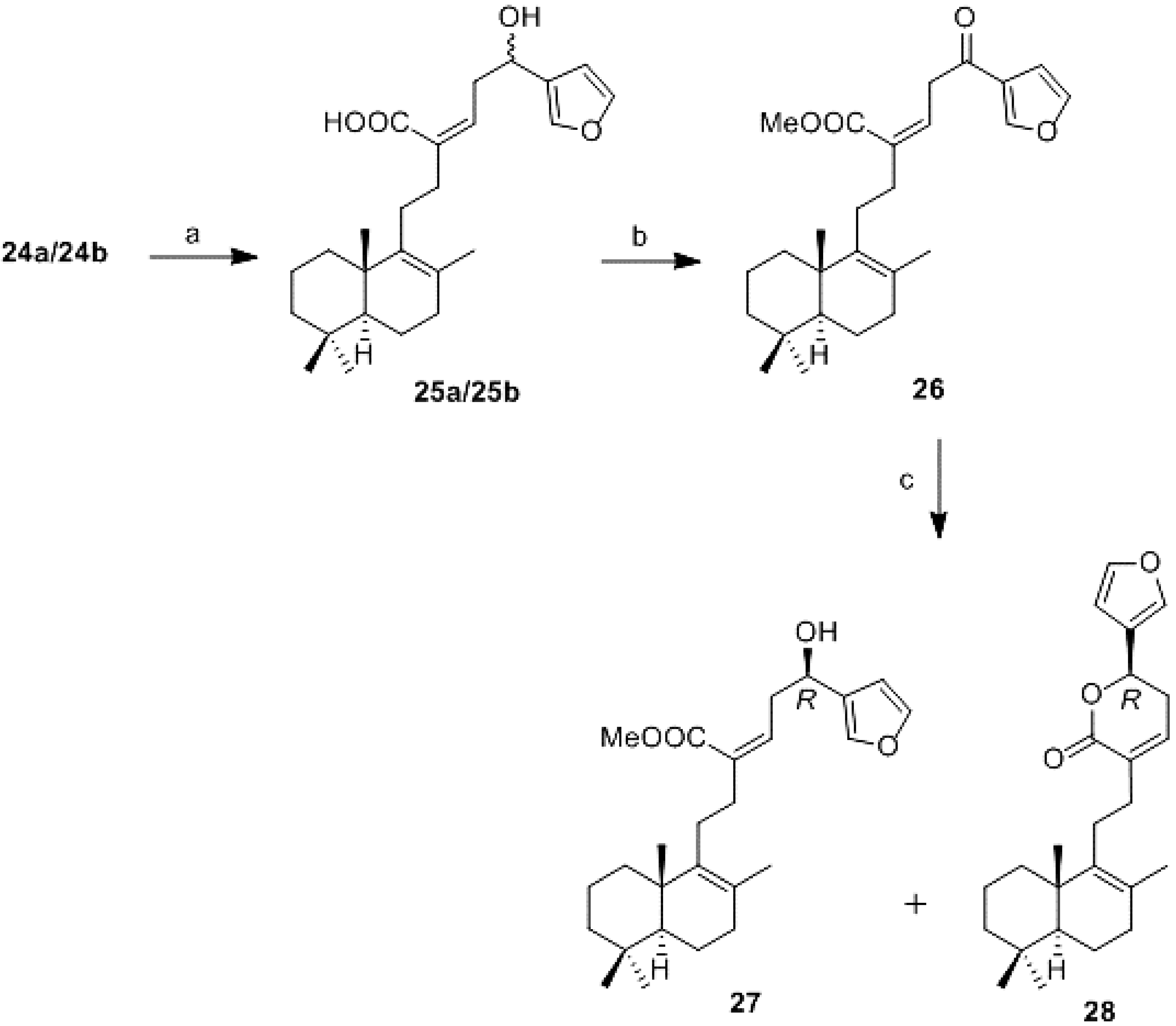
16-(R,S)-Hydroxy-19,20-epoxy-luffara-8,13Z,17(20),18-tetraen-21-oic acid (25a/25b): To a solution of 24a/24b (20 mg, 0.05 mmol) in MeOH (1.4 mL) was added anhydrous K2CO3 (14 mg, 0.1 mmol) and the mixture was reacted at rt for 7 h. Then, water and 0.01M aqueous solution of HCl were added until neutral pH was reached. It was extracted with AcOEt and the combined organic layers were washed with water until neutral pH was reached and brine, dried (Na2SO4), filtered, and concentrated in vacuo to obtain 25a/25b (20 mg, 100%). IR υ 3500–2700 (COOH), 2924, 2854, 1714 (C=O), 1456, 1377, 1261, 1026; 1H-NMR (400 MHz, CDCl3) δ 7.41 (1H, s, H-20), 7.40 (1H, s, H-19), 6.41 (1H, s, H-18), 6.08 (1H, t, J = 7.8 Hz, H-14), 4.85 (1H, t, J = 6.9 Hz, H-16), 2.95–2.85 (2H, m, H-15), 2.35–2.25 (4H, m, H-11, H-12), 2.05–1.00 (11H, m), 1.58 (3H, s, Me-22), 0.93 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.83 (3H, s, Me-24).
Methyl 16-oxo-19,20-epoxy-luffara-8,13Z,17(20),18-tetraen-21-oate (26): To a solution of 25a/25b (11 mg, 0.027 mmol) in DCM (0.5 mL) at 0 °C was added DMP (21 mg, 0.054 mmol). The reaction mixture was stirred under argon atmosphere at rt for 30 min. Then, it was added AcOEt and the organic layer was washed with 10% aqueous solution of Na2S2O3, dried (Na2SO4), filtered, and the solvent was partially removed under reduced pressure. The obtained acid was esterified directly: the crude was dissolved in C6H6/MeOH 1:1 (0.34 mL) and cooled to 0 °C. Under argon atmosphere TMSCHN2 (2.0 M in hexane; 27 μL, 0.054 mmol) was added dropwise. After 15 min the solvent was removed under reduced pressure to obtain 26 (9 mg, 89%). 1H-NMR (400 MHz, CDCl3) δ 8.14 (1H, s, H-20), 7.44 (1H, s, H-18), 6.80 (1H, s, H-19), 6.39 (t, J = 6.8 Hz, H-14), 4.05 (2H, d, J = 6.8 Hz, H-15), 3.77 (3H, s, COOMe), 2.35–2.25 (4H, m, H-11, H-12), 2.05–1.00 (11H, m), 1.57 (3H, s, Me-22), 0.93 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.83 (3H, s, Me-24).
Reduction of 26 (27 and 28): To a solution of 26 (40 mg, 0.096 mmol) in dry toluene (1.9 mL) under argon atmosphere at −78 °C, (S)-2-methyl-CBS-oxazaborolidine (1.0 M in toluene; 0.19 mL, 0.19 mmol) and borane dimethylsulfide (1.0 M in toluene; 0.19 mL, 0.19 mmol) was added. The reaction mixture was stirred at −30 °C for 20 h. It was quenched with MeOH (2 mL) and it was allowed to warm to room temperature. Then it was added water and Et2O and extracted with Et2O. The combined organic layers were washed with water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The resulting crude residue was purified with a column with Amberlyst 15 (NH4+) and after that, with a flash CC (hexane-AcOEt, 85:15) to obtain 27 (19 mg, 52%) and 28 (16 mg, 42%).
Methyl 16R-hydroxy-19,20-epoxy-luffara-8,13Z,17(20),18-tetraen-21-oate (27):
= +33.3 (c 0.2, CHCl3); IR υ 3466 (OH), 3134, 2928, 1717 (C=O),1647 (C=C), 1437, 1375, 1219, 1026; 1H-NMR (400 MHz, CDCl3) δ 7.40 (1H, s, H-20), 7.39 (1H, s, H-19), 6.41 (1H, bs, H-18), 6.00 (1H, t, J = 7.9 Hz, H-14), 4.83–4.78 (1H, m, H-16), 3.77 (3H, s, MeCOO), 2.87–2.83 (2H, m, H-15), 2.30 (2H, t, J = 8.7 Hz, H-12), 2.00–1.00 (13H, m), 1.57 (3H, s, Me-22), 0.93 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.83 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 168.9 (C), 143.2 (C), 139.7 (C), 138.9 (CH), 136.6 (CH), 135.6 (C), 128.9 (C), 126.6 (C), 108.5 (CH), 66.5 (CH), 51.8 (CH), 51.5 (CH3), 41.8 (CH2), 39.0 (C), 37.9 (CH2), 36.9 (CH2), 35.3 (CH2), 33.6 (CH2), 33.3 (C, CH3), 28.2 (CH2), 21.7 (CH3), 20.0 (CH3), 19.4 (CH3), 19.0 (CH2 - 2); HRMS (ESI) m/z calcd for C26H38O4Na (M + Na)+ 437.2662, found 437.2659.
19,20-Epoxy-luffara-8,13Z,17(20),18-tetraen-21,16R-olide (28):
= +56.0 (c 0.26, CHCl3); IR υ 2924, 2855, 1724 (C=O), 1464, 1377, 1117; 1H-NMR (400 MHz, CDCl3) δ 7.49 (1H, s, H-20), 7.42 (1H, s, H-19), 6.46 (1H, bs, H-18), 6.65–6.60 (1H, m, H-14), 5.39 (1H, dd, J = 4.1 and 11.0 Hz, H-16), 3.15–3.00 (2H, m, H-15), 2.50–1.00 (15H, m), 1.62 (3H, s, Me-22), 0.95 (3H, s, Me-23), 0.89 (3H, s, Me-25), 0.84 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 165.0 (C), 143.6 (CH), 139.9 (CH), 139.5 (C), 137.2 (CH), 133.8 (C), 126.9 (C), 124.0 (C), 108.6 (CH), 72.3 (CH), 51.9 (CH), 41.8 (CH2), 39.0 (C), 37.0 (CH2), 33.6 (CH2), 33.3 (C, CH3), 31.6 (CH2), 30.5 (CH2), 27.7 (CH2), 21.7 (CH3), 20.1 (CH3), 19.5 (CH3), 19.0 (CH2-2); HRMS (ESI) m/z calcd for C25H34O3Na (M + Na)+ 405.2400, found 405.2405.
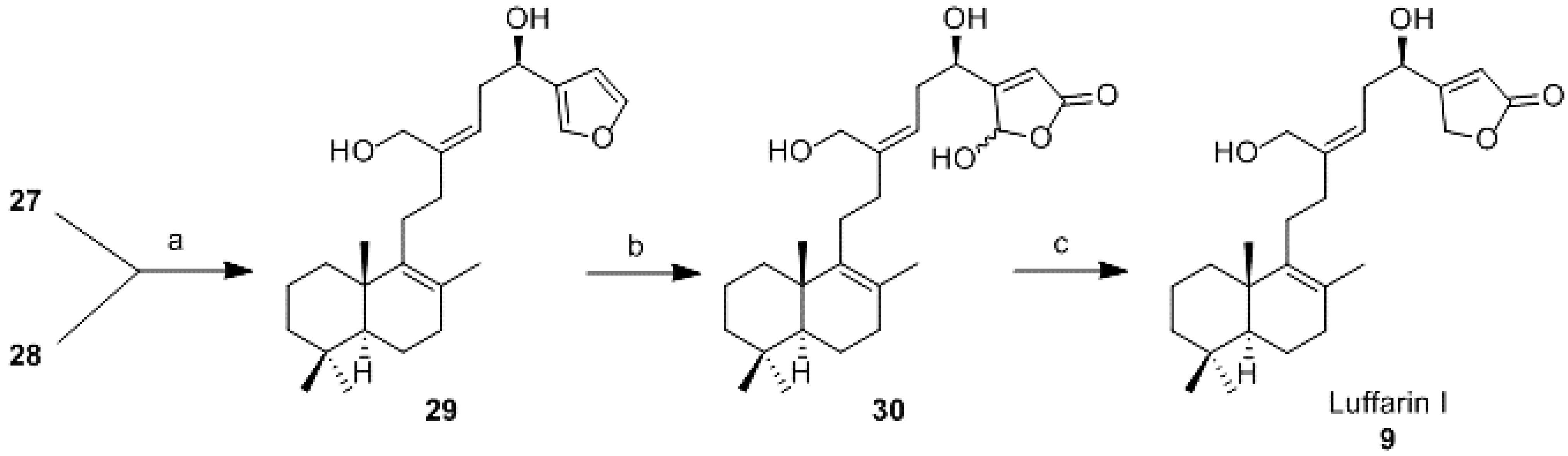
Reduction of 27 and 28 (29): To a solution of 27 (14 mg, 0.034 mmol) in DCM (0.3 mL) under argon atmosphere, DIBAL-H (1.0 M in hexane; 0.2 mL, 0.2 mmol) was added dropwise. The mixture was reacted at rt for 90 min and then, AcOEt was added. It was quenched with a saturated aqueous solution of potassium sodium tartrate (1 mL), and it was stirred for 15 min. After that it was extracted with AcOEt and the combined organic layers were washed with 6% aqueous solution of NaHCO3, water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The resulting crude residue was purified by CC (hexane-AcOEt 9:1) to obtain 29 (11 mg, 85%).
To a solution of 28 (3 mg, 0.008 mmol) in DCM (0.22 mL) under argon atmosphere, DIBAL-H (1.0 M in hexane; 48 μL, 0.05 mmol) was added dropwise. The mixture was reacted at rt for 2.5 h and then, AcOEt was added. It was quenched with a saturated aqueous solution of potassium sodium tartrate (1 mL), and it was stirred for 15 min. After that it was extracted with AcOEt and the combined organic layers were washed with 6% aqueous solution of NaHCO3, water and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The resulting crude residue was purified by CC (hexane-AcOEt 9:1) to obtain 29 (3 mg, 95%).
19,20-Epoxy-luffara-8,13Z,17(20),18-tetraene-16R,21-diol (29):
= +51.2 (c 0.37, CHCl3); IR υ 3345 (OH), 2928, 2866, 1456, 1161, 1024; 1H-NMR (400 MHz, CDCl3) δ 7.40 (2H, s, H-19, H-20), 6.41 (1H, s, H-18), 5.41 (1H, t, J = 7.9 Hz, H-14), 4.73 (1H, dd, J = 4.4 and 8.0 Hz, H-16), 4.20 (1H, d, J = 11.6 Hz, HA-21), 4.05 (1H, d, J = 11.6 Hz, HB-21), 2.60–2.50 (2H, m, H-15), 2.20–2.10 (4H, m, H-11, H-12), 2.00–1.00 (11H, m), 1.58 (3H, s, Me-22), 0.95 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.83 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 144.0 (C), 143.3 (CH), 140.1 (C), 138.9 (CH), 128.7(C), 126.1 (C), 123.0 (CH), 108.5 (CH), 66.0 (CH), 60.2 (CH2), 51.9 (CH), 41.8 (CH2), 39.0 (C), 37.1 (CH2), 37.0 (CH2), 36.3 (CH2), 33.6 (CH2), 33.3 (C, CH3), 27.4 (CH2), 21.7 (CH3), 20.1 (CH3), 19.5 (CH3), 19.0 (CH2-2); HRMS (ESI) m/z calcd for C25H38O3Na (M + Na)+ 409.2713, found 409.2695.
16R,20(R,S),21-Trihydroxy-luffara-8,13Z,17-trien-19,20-olide (30): To a solution of 29 (15 mg, 0.038 mmol) in DCM (5.6 mL), DIPEA (74 μL, 0.38 mmol) and Bengal Rose (1 mg) were added. After that, anhydrous oxygen was bubbled in for 10 min, the mixture was cooled to −78 °C and under oxygen atmosphere, it was irradiated by 200 W light for 5 h. Then, it was allowed to warm to room temperature and oxalic acid (5 mL) was added. The mixture was stirred for 30 min. Afterwards water was added and the mixture was extracted with DCM. The combined organic layers were washed with water, dried (Na2SO4), filtered, and concentrated in vacuo. The resulting crude residue was purified by flash CC (hexane-AcOEt; 1:1) to afford 30 (16 mg, 99%); IR υ 3308 (OH), 2924, 2851, 1748 (C=O), 1456, 1261, 1024; 1H-NMR (400 MHz, CDCl3) δ 6.17 (1H, s, H-20), 6.03 (1H, s, H-18), 5.45–5.35 (1H, m, H-14), 4.80–4.70 (1H, m, H-16), 4.25–4.00 (2H, m, H-21), 2.65-2.55 (2H, m, H-15), 2.20–2.10 (4H, m, H-11, H-12), 2.00–1.00 (11H, m), 1.56 (3H, s, Me-22), 0.94 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.83 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 167.8 (C), 167.7 (C), 144.0(C), 139.8 (C), 126.2 (C), 122.5 (CH), 117.8 (CH), 98.0 (CH), 68.1 (CH), 60.0 (CH2), 51.9 (CH), 42.0 (CH2), 39.0 (C), 37.4 (CH2), 36.7 (CH2), 33.7 (CH2), 33.6 (CH2), 33.3 (C, CH3), 27.2 (CH2), 21.7 (CH3), 20.1 (CH3), 19.5 (CH3), 19.0 (CH2-2).
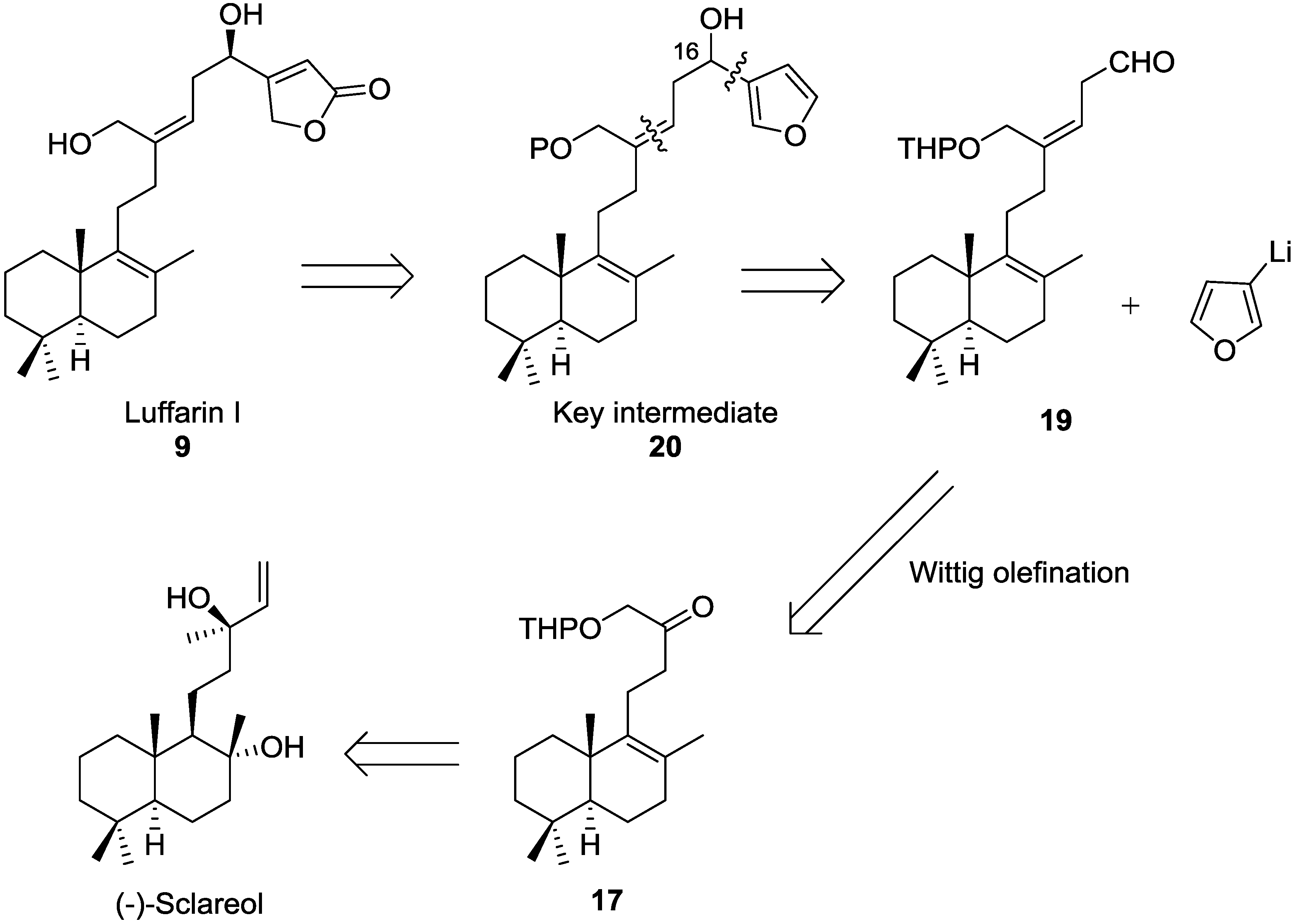
16R,21-Dihydroxy-luffara-8,13Z,17-trien-19,20-olide (Luffarin I (9)): To a solution of 30 (7 mg, 0.017 mmol) in absolute ethanol (1.1 mL) at 0 °C, NaBH4 (2 mg, 0.061 mmol) was added. After 5 min, water and 2 M aqueous solution of HCl was added. It was extracted with AcOEt and the combined organic layers were washed with water until neutral pH was reached and brine, dried (Na2SO4), filtered, and concentrated in vacuo. The resulting crude residue was purified by CC (hexane-AcOEt; 1:1) to obtain 9 (5 mg, 71%).
= +69.0 (c 0.51, CHCl3); IR υ 3387 (OH), 2926, 2866, 1780 (C=O), 1748, 1638 (C=C), 1456, 1026; 1H-NMR (400 MHz, CDCl3) δ 5.99 (1H, bs, H-18), 5.40 (1H, t, J = 8.1 Hz, H-14), 4.89 (2H, bs, H-20), 4.66 (1H, t, J = 5.9 Hz, H-16), 4.22 (1H, d, J = 11.6 Hz, HA-21), 4.14 (1H, d, J = 11.6 Hz, HB-21), 2.60–2.50 (2H, m, H-15 ), 2.20–2.00 (4H, m, H-11, H-12), 2.00–1.00 (11H, m), 1.57 (3H, s, Me-22), 0.94 (3H, s, Me-23), 0.88 (3H, s, Me-25), 0.83 (3H, s, Me-24); 13C-NMR (100 MHz, CDCl3) δ 173.6 (C), 172.3 (C), 145.1 (C), 139.7 (C), 126.4 (C), 121.8 (CH), 114.9 (CH), 71.3 (CH2), 67.4 (CH), 60.3 (CH2), 51.8 (CH), 41.7 (CH2), 39.0 (C), 37.6 (CH2), 37.0 (CH2), 35.1 (CH2), 33.6 (CH2), 33.3 (C, CH3), 27.3 (CH2), 21.6 (CH3), 20.1 (CH3), 19.5 (CH3), 19.0 (CH2 - 2); HRMS (ESI) m/z calcd for C25H38O4Na (M + Na)+ 425.2662, found 425.2671.
Supplementary files available. Copies of IR, HRMS, NMR spectra and study of C-16 stereochemistry of compound 29 using Mosher’s methodology are included.



 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}