3. Experimental Section
IR spectra were obtained with a Shimadzu Prestige 21/IRAffinity-1 FT-IR spectrometer. 1H- and 13C-NMR spectra were recorded on a JEOL JNM-ECA 500 FT NMR spectrometer at 500 MHz for 1H and 125 MHz for 13C; a JEOL JNM-AL 400 NMR spectrometer at 400 MHz for 1H and 100 MHz for 13C; and a JEOL JNM-AL 300 NMR spectrometer at 300 MHz for 1H and 75 MHz for 13C (ppm, J in Hz with TMS as internal standard). All proton and carbon signals were assigned by extensive NMR measurements using COSY, HMBC, and HMQC techniques. Mass spectra were recorded on a JEOL JMS 700 instrument with a direct inlet system operating at 70 eV. Elemental analyses were conducted on a YANACO MT-6 CHN CORDER elemental analyzer.
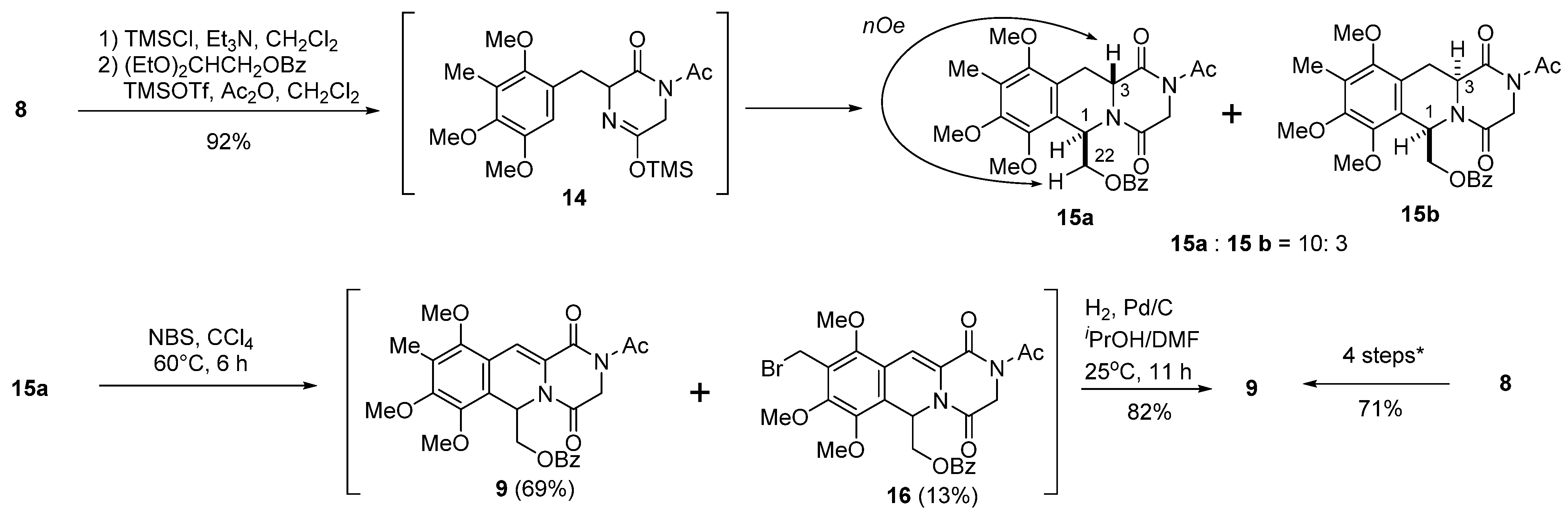
3.1. ((6R*,11aR*)-2-Acetyl-7,8,10-trimethoxy-9-methyl-1,4-dioxo-1,3,4,6,11,11a-hexahydro-2H-pyrazino-(1,2-b)isoquinolin-6-yl)methyl Benzoate (15a) and ((6R*,11aS*)-2-acetyl-7,8,10-trimethoxy-9-methyl- 1,4-dioxo-1,3,4,6,11,11a-hexahydro-2H-pyrazino(1,2-b)isoquinolin-6-yl)methyl Benzoate (15b)
TMSCl (498 μL, 3.9 mmol) was added to a stirred solution of
8 (1.05 g, 3.0 mmol) in dichloromethane (18 mL) and TEA (544 μL, 3.9 mmol), and stirring was continued at 25 °C for 2 h. A solution of 2,2-diethoxyethyl benzoate [
28] (785.8 mg, 3.3 mmol) in dichloromethane (12 mL) followed by TMSOTf (2.71 mL, 15 mmol) was added dropwise for 5 min each, and then Ac
2O (283.6 μL, 3.0 mmol) was added in one portion at 25 °C and the reaction mixture was stirred for 4 h. The reaction mixture was diluted with saturated NaHCO
3 solution (100 mL) and extracted with CHCl
3 (100 mL × 3). The combined extracts were washed with brine (100 mL), dried, and concentrated
in vacuo. The residue was subjected to column chromatography with ethyl acetate–hexane (1:2) to give
15 (1.37 g, 92%,
15a:
15b = 10:3) as a colorless amorphous powder, which was an inseparable mixture of diastereomers. Each authentic sample was obtained by chromatography on preparative layer silica gel plates (Merck 5715).
Compound 15a: 1H-NMR (400 MHz, CDCl3) δ: 7.99 (2H, m, Ar-H), 7.57 (1H, m, Ar-H), 7.44 (2H, m, Ar-H), 6.20 (1H, dd, J = 9.5, 3.9 Hz, C6-H), 4.75 (1H, dd, J = 11.7, 9.5 Hz, C12-H), 4.70 (1H, dd, J = 9.8, 5.4 Hz, C11a-H), 4.52 (1H, dd, J = 11.7, 3.9 Hz, C12-H), 4.36 (1H, d, J = 18.0 Hz, C3-H), 4.31 (1H, d, J = 18.0, C3-H), 3.95 (3H, s, C7-OMe), 3.80 (3H, s, C8-OMe), 3.72 (3H, s, C10-OMe), 3.40 (1H, dd, J = 16.6, 5.4 Hz, C11-Hα), 3.14 (1H, dd, J = 16.6, 9.8 Hz, C11-Hβ), 2.59 (3H, s, COCH3), 2.21 (3H, s, C9-Me). 13C-NMR (100 MHz, CDCl3) δ: 171.7 (s, COCH3), 167.9 (s, C1), 166.6 (s, OCOPh), 163.2 (s, C4), 152.4 (s, C10), 150.6 (s, C8), 146.2 (s, C7), 133.3 (d, Ph), 129.7 (d, Ph × 2), 129.6 (s, Ph), 128.5 (d, Ph × 2), 125.9 (s, C9), 122.6 (s, C6a), 121.4 (s, C10a), 63.6 (t, C12), 60.6 (q, C7-OCH3), 60.3 (q, C10-OCH3), 60.0 (q, C8-OCH3), 54.0 (d, C11a), 48.3 (d, C6), 45.7 (t, C3), 27.2 (q, COCH3), 26.0 (t, C11), 9.5 (q, C9-CH3). FT-IR (KBr) cm−1: 1717, 1686, 1672, 1275, 714. EI-MS m/z (%): 496 (M+, 11), 361 (100), 319 (13), 234 (15), 204 (8), 105 (7). HR-EI-MS: calcd for C26H28N2O8, 496.1846, found: 496.1841.
Compound 15b: 1H-NMR (400 MHz, CDCl3) δ: 7.93 (2H, m, Ar-H), 7.54 (1H, m, Ar-H), 7.42 (2H, m, Ar-H), 6.15 (1H, dd, J = 7.0, 4.6 Hz, C6-H), 5.10 (1H, d, J = 16.0 Hz, C3-H), 4.55 (1H, dd, J = 11.5, 7.0 Hz, C12-H), 4.37 (1H, dd, J = 11.5, 4.6 Hz, C12-H), 4.07 (1H, dd, J = 12.2, 4.9 Hz, C11a-H), 3.94 (3H, s, C7-OMe), 3.79 (3H, s, C8-OMe), 3.78 (1H, d, J =16.0 Hz, C3-H), 3.68 (1H, dd, J =15.9, 4.9 Hz, C11-Hα), 3.64 (3H, s, C10-OMe), 3.05 (1H, dd, J = 15.9, 12.2 Hz, C11-Hβ), 2.59 (3H, s, COMe), 2.22 (3H, s, C9-Me). 13C-NMR (100 MHz, CDCl3) δ: 170.9 (s, COCH3), 168.7 (s, C1), 166.4 (s, OCOPh), 166.1 (s, C4), 151.7 (s, C10), 150.6 (s, C8), 146.2 (s, C7), 133.2 (d, Ph × 2), 129.7 (s, Ph), 129.6 (d, Ph), 128.4 (d, Ph × 2), 126.1 (s, C9), 123.6 (s, C6a), 121.8 (s, C10a), 66.1 (t, C12), 61.0 (q, C10-OCH3), 60.7 (q, C7-OCH3), 60.0 (q, C8-OCH3), 56.3 (d, C11a), 48.5 (d, C6), 45.6 (t, C3), 26.9 (q, COCH3), 22.8 (t, C11), 9.4 (q, C9-CH3). FT-IR (KBr) cm−1: 1721, 1707, 1692, 1412, 1368, 1273. EI-MS m/z (%): 496 (M+, 10), 361 (100), 319 (11), 234 (13), 204 (7), 105 (7). HR-EI-MS: calcd for C26H28N2O8, 496.1846, found: 496.1841.
3.2. (2-Acetyl-7,8,10-trimethyl-1,4-dioxo-1,3,4,6-tetrahydro-2H-pyrazino(1,2-b)Isoquinoline-6-yl)methyl Beozoate (9) and (2-acetyl-9-(bromomethyl)-,7,8,10-trimethoxy-1,4-dioxo-1,3,4,6-tetrahydro-2H-pyrazino(1,2-b)isoquinolin-6-yl)methyl Beozoate (16)
NBS (106.8 mg, 0.6 mmol) was added to a stirred solution of 15a (150.0 mg, 0.3 mmol) in dry CCl4, and the reaction mixture was heated at 60 °C for 6 h. The reaction mixture was filtered through a short pad of Celite, and the filtrate was washed with CCl4. The combined filtrates were concentrated in vacuo, and the residue was purified by flash chromatography on silica gel with ethyl acetate–hexane (1:3) as solvent to give 9 (102.0 mg, 69%) and 16 (17.7 mg, 13%).
Compound 9: 1H-NMR (400 MHz, CDCl3) δ: 7.91 (2H, m, Ar-H), 7.55 (1H, m, Ar-H), 7.47 (1H, s, C11-H), 7.42 (2H, m, Ar-H), 6.47 (1H, dd, J = 7.7, 3.8 Hz, C6-H), 4.93 (1H, d, J = 17.6 Hz, C3-H), 4.52 (1H, dd, J = 11.7, 7.7 Hz, C12-H), 4.27 (1H, dd, J = 11.7, 3.8 Hz, C12-H), 3.96 (3H, s, C7-OMe), 3.92 (1H, d, J =17.6 Hz, C3-H), 3.85 (3H, s, C8-OMe), 3.71 (3H, s, C10-OMe), 2.61 (3H, s, COMe), 2.21 (3H, s, C9-Me). 13C-NMR (100 MHz, CDCl3) δ: 171.8 (s, COCH3), 166.3 (s, OCOPh), 162.7 (s, C4), 160.7 (s, C1), 154.1 (s, C8), 152.4 (s, C10), 146.0 (s, C7), 133.3 (d, Ph), 129.6 (d, Ph × 2), 129.4 (s, Ph), 128.5 (d, Ph × 2), 126.7 (s, C9), 125.8 (s, C11a or C10a), 121.5 (s, C6a), 118.9 (s, C11a or C10a), 114.7 (d, C11), 64.3 (t, C12), 62.1 (q, C10-OCH3), 60.8 (q, C7-OCH3), 60.2 (q, C8-OCH3), 47.9 (d, C6), 45.2 (t, C3), 26.7 (q, COCH3), 9.4 (q, C9-CH3). FT-IR (KBr) cm−1: 1701, 1416, 1393, 1368, 1269, 1211, 1088, 1072, 1043, 1028, 1007, 964, 459. EI-MS m/z (%): 494 (M+, 14), 359 (100), 260 (84), 232 (38), 217 (6), 202 (5), 105 (6). HR-EI-MS: calcd for C26H26N2O8, 494.1689, found: 494.1686.
Compound 16: 1H-NMR (400 MHz, CDCl3) δ: 7.90 (2H, m, Ar-H), 7.55 (1H, m, Ar-H), 7.42 (2H, m, Ar-H), 7.41 (1H, s, C11-H), 6.49 (1H, dd, J = 7.7 Hz, 3.8 Hz, C6-H), 4.94 (1H, d, J =17.6 Hz, C3-H), 4.60 (1H, d, J =9.1 Hz, C9-CH2Br), 4.57 (1H, d, J = 9.1, C9-CH2Br), 4.55 (1H, dd, J = 11.6 Hz, 7.7 Hz, C12-H), 4.30 (1H, dd, J = 11.6 Hz, 3.8 Hz, C12-H), 4.03 (3H, s, C8-OMe), 3.97 (3H, s, C7-OMe), 3.90~3.95 (1H, overlapped, C3-H), 3.90 (3H, s, C10-OMe), 2.62 (3H, s, COCH3). 13C-NMR (100 MHz, CDCl3) δ: 171.8 (s, COCH3), 166.3 (s, OCOPh), 162.7 (s, C4), 160.4 (s, C1), 153.9 (s, C8), 152.1 (s, C10), 146.0 (s, C7), 133.4 (d, Ph), 129.6 (d, Ph × 2), 129.3 (s, Ph), 128.5 (d, Ph × 2), 127.3 (s, C9), 126.3 (s, C10a or C11a), 125.1 (s, C6a), 119.4 (s, C10a or C11a), 113.6 (d, C11), 64.2 (t, C12), 63.4 (q, C10-OCH3), 60.7 (q, C7-OCH3 and C8-OCH3), 47.9 (d, C6), 45.2 (t, C3), 26.7 (q, COCH3), 21.5 (t, C9-CH2Br). FT-IR (KBr) cm−1: 1701, 1368, 1292, 1269, 1215. EI-MS m/z (%): 572 (M+, 15), 493 (9), 439 (100), 417 (14), 393 (13), 375 (5), 371 (8), 364 (8), 359 (24), 339 (11), 310 (21), 274 (9), 260 (27), 231 (10), 105 (21). HR-EI-MS: calcd for C26H25BrN2O8, 572.0794, found: 572.0796.
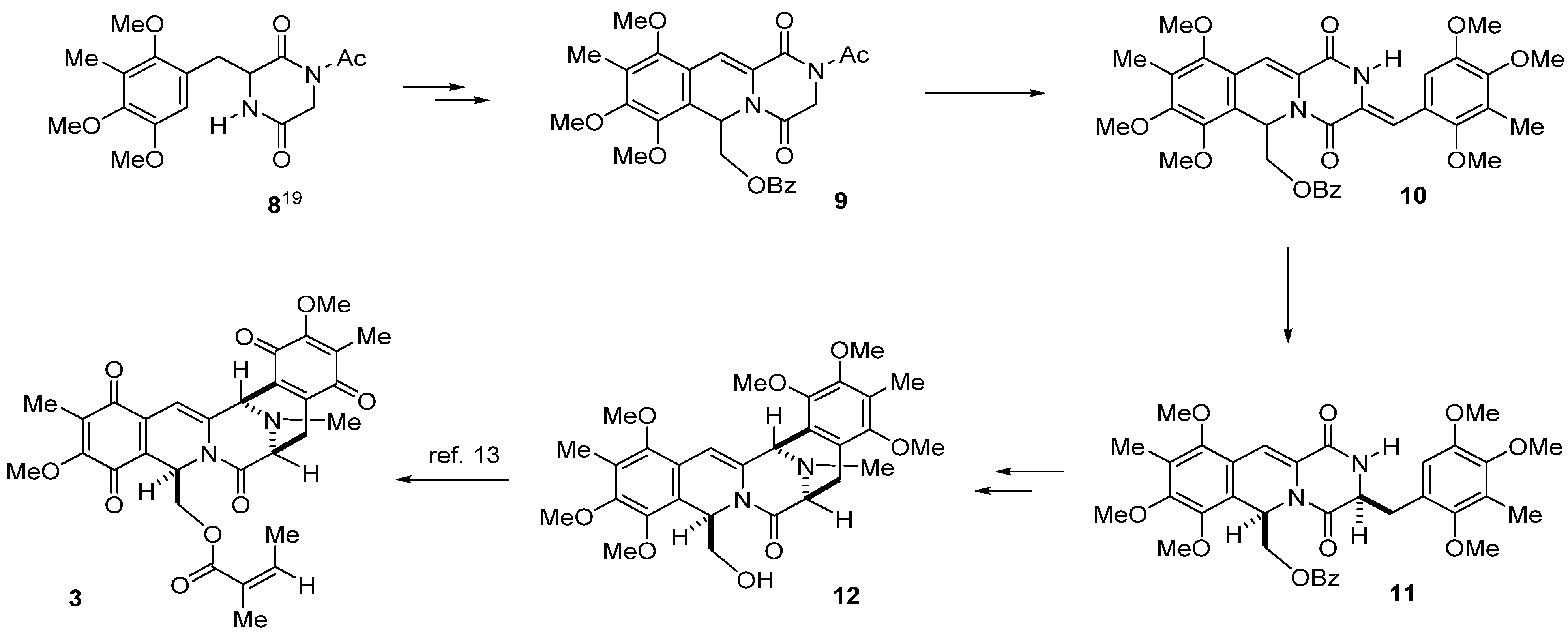
3.3. (2-Acetyl-7,8-10-trimethyl-1,4-dioxo-1,3,4,6-tetrahydro-2H-pyrazino(1,2-b)isoquinolin-6-yl)methyl Benzoate (9) from 8 in Four Steps
TMSCl (498 μL, 3.9 mmol) was added to a stirred solution of 8 (1.05 g, 3.0 mmol) in dichloromethane (18 mL) and TEA (544 μL, 3.9 mmol), and stirring was continued at 25 °C for 2 h. A solution of 2,2-diethoxyethyl benzoate (785.8 mg, 3.3 mmol) in dichloromethane (12 mL) followed by TMSOTf (2.71 mL, 15 mmol) was added dropwise respectively over 5 min. Then, Ac2O (283.6 μL, 3.0 mmol) was added at 25 °C and the reaction mixture was stirred for 4 h. The reaction mixture was diluted with saturated NaHCO3 solution (100 mL) and extracted with CHCl3 (100 mL × 3). The combined extracts were washed with brine (100 mL), dried, and concentrated in vacuo. The residue was subjected to column chromatography with ethyl acetate–hexane (1:2) to give 15 (1.37 g, 92%, 15a:15b = 10:3) as a colorless amorphous powder. Diastereomeric mixture 15 was dissolved in CCl4 (90 mL) and NBS (106.8 mg, 5.46 nmol) was added at 25 °C, and the reaction mixture was heated at 60 °C for 3 h. The reaction mixture was filtered through a short pad of Celite, and the filtrate was washed with CCl4. The combined filtrates were concentrated in vacuo, and the residue was used in the next step without further purification. The above residue was dissolved in 2-propanol/ DMF (1:0.2) (72 mL) and was hydrogenated over 10% Pd/C (980 mg) at 25 °C for 11 h. The catalyst was removed by filtration and washed with CHCl3 and MeOH. The combined filtrates were diluted with H2O (100 mL) and extracted with CHCl3 (100 mL × 3). The combined extracts were washed with brine (100 mL), dried, and concentrated in vacuo. The residue was subjected to column chromatography with ethyl acetate–hexane (1:4) to give 9 (1.05 g, 71% overall yield, 4 steps) as a colorless amorphous powder.
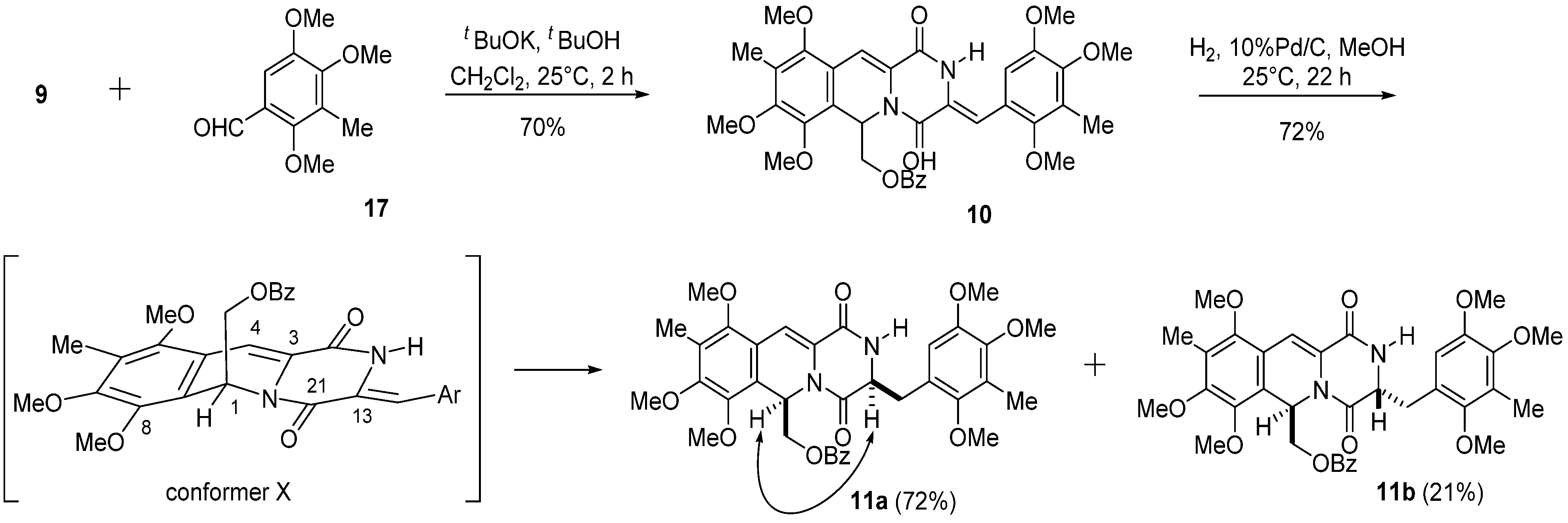
3.4. (Z)-(7,8,10-Trimethoxy-9-methyl-1,4-dioxo-3-(2,4,5-trimethoxy-3-methylbenzylidene)-1,3,4,6-tetra-hydro-2H-pyrazino(1,2-b)isoquinolin-6-yl)methyl Benzoate (10)
A solution of t-BuOK in t-BuOH (1 M, 2.4 mL, 2.4 mmol) was added to a solution of 9 (988 mg, 2.0 mmol) and 2,4,5-trimethoxybenzaldehyde (17) (420 mg, 2.0 mmol) in CH2Cl2 (20 mL) over 1 h at 0 °C, and the reaction mixture was stirred at 25 °C for 2 h. The reaction mixture was diluted with saturated NH4Cl (100 mL) and extracted with CH2Cl2 (100 mL × 3). The combined extracts were washed with brine (100 mL), dried, and concentrated in vacuo. The residue was subjected to column chromatography with ethyl acetate–hexane (1:2) to give 10 (901 mg, 70%) as a pale yellow amorphous powder.
Compound 10: 1H-NMR (400 MHz, CDCl3) δ: 9.40 (1H, s, N-H), 7.93 (2H, m, Ar-H), 7.49 (1H, m, Ar-H), 7.36 (2H, m, Ar-H), 7.31 (1H, s, C11-H), 6.94 (1H, s, C3a-H), 6.68 (1H, dd, J = 6.8, 3.9 Hz, C6-H), 6.62 (1H, s, C6′-H), 4.52 (1H, dd, J = 11.7, 6.8 Hz, C12-H), 4.45 (1H, dd, J = 11.7, 3.9 Hz, C12-H), 3.98 (3H, s, C7-OMe), 3.84 (3H, s, C8-OMe), 3.83 (3H, s, C4′-OMe), 3.83 (3H, s, C5′-OMe), 3.69 (3H, s, C10-OMe), 3.58 (3H, s, C2′-OMe), 2.24 (3H, s, C3′-Me), 2.19 (3H, s, C9-Me). 13C-NMR (100 MHz, CDCl3) δ: 166.1 (s, OCOPh), 157.4 (s, C4), 156.5 (s, C1), 153.3 (s, C8), 151.8 (s, C10), 149.6 (s, C5′), 149.2 (s, C2′), 149.0 (s, C4′), 146.0 (s, C7), 132.9 (d, Ph), 129.7 (s, Ph), 129.7 (d, Ph × 2), 128.2 (d, Ph × 2), 126.6 (s, C3′), 126.4 (s, C9), 126.2 (s, C11a), 125.4 (s, C3), 121.6 (s, C1′), 120.6 (s, C6a), 119.1 (s, C10a), 114.0 (d, C3a), 111.9 (d, C6′), 110.8 (d, C11), 65.0 (t, C12), 62.0 (q, C10-OCH3), 60.9 (q, C2′-OCH3), 60.7 (q, C7-OCH3), 60.4 (q, C4′-OCH3), 60.1 (q, C8-OCH3), 55.9 (q, C5′-OCH3), 48.7 (d, C6), 9.5 (q, C3′-CH3), 9.3 (q, C9-CH3). FT-IR (KBr) cm−1: 3246, 1724, 1688, 1628, 1489, 1468, 1452, 1414, 1387, 1369, 1323, 1269, 1248, 1090, 1067, 1003, 712. EI-MS m/z (%): 644 (M+, 7), 509 (100), 481 (5), 232 (10). HR-EI-MS: calcd for C35H36N2O10, 644.2370, found: 644.2369.
3.5. ((3S*,6R*)-7,8,10-Trimethoxy-9-methyl-1,4-dioxo-3-(2,4,5-trimethoxy-3-methylbenzyl)-1,3,4,6-tetrahydro-2H-pyrazino(1,2-b)isoquinolin-6-yl)methyl Benzoate (11a) and ((3R*,6R*)-7,8,10-trimethoxy-9-methyl-1,4-dioxo-3-(2,4,5-trimethoxy-3-methylbenzyl)-1,3,4,6-tetrahydro-2H-pyrazino-(1,2-b)isoquinolin-6-yl)methyl Benzoate (11b)
A solution of 10 (644.0 mg, 1.0 mmol) in MeOH (50 mL) was hydrogenated over 10% Pd/C (213.0 mg) at 25 °C for 22 h. The catalyst was removed by filtration and washed with CHCl3 and MeOH. The combined filtrate was concentrated in vacuo. The residue was subjected to column chromatography with ethyl acetate–hexane (1:2) to give 11b (137.0 mg, 21%) as a pale yellow amorphous powder, and with ethyl acetate–hexane (2:1) to give 11a (463.0 mg, 72%) as a pale yellow amorphous powder.
Compound 11a: 1H-NMR (400 MHz, CDCl3) δ: 8.00 (2H, m, Ar-H), 7.53 (1H, m, Ar-H), 7.42 (2H, m, Ar-H), 7.25 (1H, s, C11-H), 6.53 (1H, dd, J = 7.5, 4.2 Hz, C6-H), 6.51 (1H, s, C6′-H), 6.22 (1H, s, N-H), 4.45 (1H, dd, J = 11.4, 7.5 Hz, C12-H), 4.34 (1H, dd, J = 10.3, 3.3 Hz, C3-H), 4.31 (1H, dd, J =11.4, 4.2 Hz, C12-H), 3.97 (3H, s, C7-OMe), 3.82 (3H, s, C8-OMe), 3.80 (3H, s, C5′-OMe), 3.78 (3H, s, C4′-OMe), 3.67 (3H, s, C10-OMe), 3.64 (3H, s, C2′-OMe), 3.52 (1H, dd, J =13.8, 3.3 Hz, C3a-H), 2.73 (1H, dd, J =13.8, 10.3 Hz, C3a-H), 2.20 (3H, s, C3′-Me), 2.19 (3H, s, C9-Me). 13C-NMR (100 MHz, CDCl3) δ: 166.1 (s, OCOPh), 165.4 (s, C4), 160.7 (s, C1), 153.2 (s, C8), 151.8 (s, C10), 150.9 (s, C2′), 149.7 (s, C5′), 147.6 (s, C4′), 146.0 (s, C7), 133.1 (d, Ph), 129.9 (d, Ph × 2), 129.8 (s, Ph), 128.3 (d, Ph × 2), 126.4 (s, C9), 126.3 (s, C3′), 126.3 (s, C11a), 123.7 (s, C1′), 121.2 (s, C6a), 119.1 (s, C10a), 111.5 (d, C6′), 110.7 (d, C11), 64.6 (t, C12), 62.0 (q, C10-OCH3), 60.8 (q, C7-OCH3), 60.6 (q, C2′-OCH3), 60.2 (q, C4′-OCH3), 60.1 (q, C8-OCH3), 56.0 (q, C5′-OCH3) 55.3 (d, C3), 47.8 (d, C6), 33.5 (t, C3a), 9.7 (q, C3′-CH3), 9.4 (q, C9-CH3).
FT-IR (KBr) cm−1: 3391, 1724, 1690, 1628, 1489, 1468, 1458, 1414, 1368, 1337, 1269, 1240, 1121, 1088. EI-MS m/z (%): 646 (M+, 14), 511 (100), 509 (13), 483 (36), 260 (8), 232 (17), 195 (10). HR-EI-MS: calcd for C35H38N2O10, 646.2527, found: 646.2523.
Compound 11b: 1H-NMR (400 MHz, CDCl3) δ: 7.93 (2H, m, Ar-H), 7.52 (1H, m, Ar-H), 7.39 (2H, m, Ar-H), 6.91 (1H, s, C11-H), 6.43 (1H, dd, J = 7.1, 4.4 Hz, C6-H), 6.39 (1H, s, C6′-H), 4.45 (1H, dd, J = 8.9, 5.2 Hz, C3-H), 4.36 (1H, dd, J = 11.7, 7.1 Hz, C12-H), 4.31 (1H, dd, J =11.7, 4.4 Hz, C12-H), 3.98 (3H, s, C7-OMe), 3.83 (3H, s, C8-OMe), 3.66 (3H, s, C4′-OMe), 3.62 (3H, s, C5′-OMe), 3.61 (3H, s, C10-OMe), 3.57 (3H, s, C2′-OMe), 3.08 (1H, dd, J = 14.5, 8.9 Hz, C3a-H), 3.06 (1H, dd, J = 14.5, 5.2 Hz, C3a-H), 2.17 (3H, s, C9-Me), 2.05 (3H, s, C3′-Me). 13C-NMR (100 MHz, CDCl3) δ: 166.2 (s, OCOPh), 165.0 (s, C4), 160.6 (s, C1), 153.0 (s, C8), 151.5 (s, C10), 151.5 (s, C2′), 149.1 (s, C5′), 147.7 (s, C4′), 145.9 (s, C7), 133.0 (d, Ph), 129.7 (s, Ph), 129.7 (d, Ph × 2), 128.3 (d, Ph × 2), 126.2 (s, C9), 125.8 (s, C3′), 125.4 (s, C11a or C10a), 121.9 (s, C1′), 120.7 (s, C6a), 119.0 (s, C11a or C10a), 112.0 (d, C6′), 109.3 (d, C11), 64.9 (t, C12), 61.9 (q, C10-OCH3), 60.7 (q, C7-OCH3), 60.5 (q, C2′-OCH3), 60.2 (q, C4′-OCH3), 60.1 (q, C8-OCH3), 57.0 (d, C3), 55.8 (q, C5′-OCH3), 47.9 (d, C6), 36.5 (t, C3a), 9.2 (q, C9-CH3 and C3′-CH3). FT-IR (KBr) cm−1: 3300, 1724, 1692, 1628, 1487, 1468, 1414, 1379, 1321, 1271, 1119, 1088. EI-MS m/z (%): 646 (M+, 11), 511 (100), 483 (39), 232 (16), 195 (9). HR-EI-MS: calcd for C35H38N2O10, 646.2527, found: 646.2521.
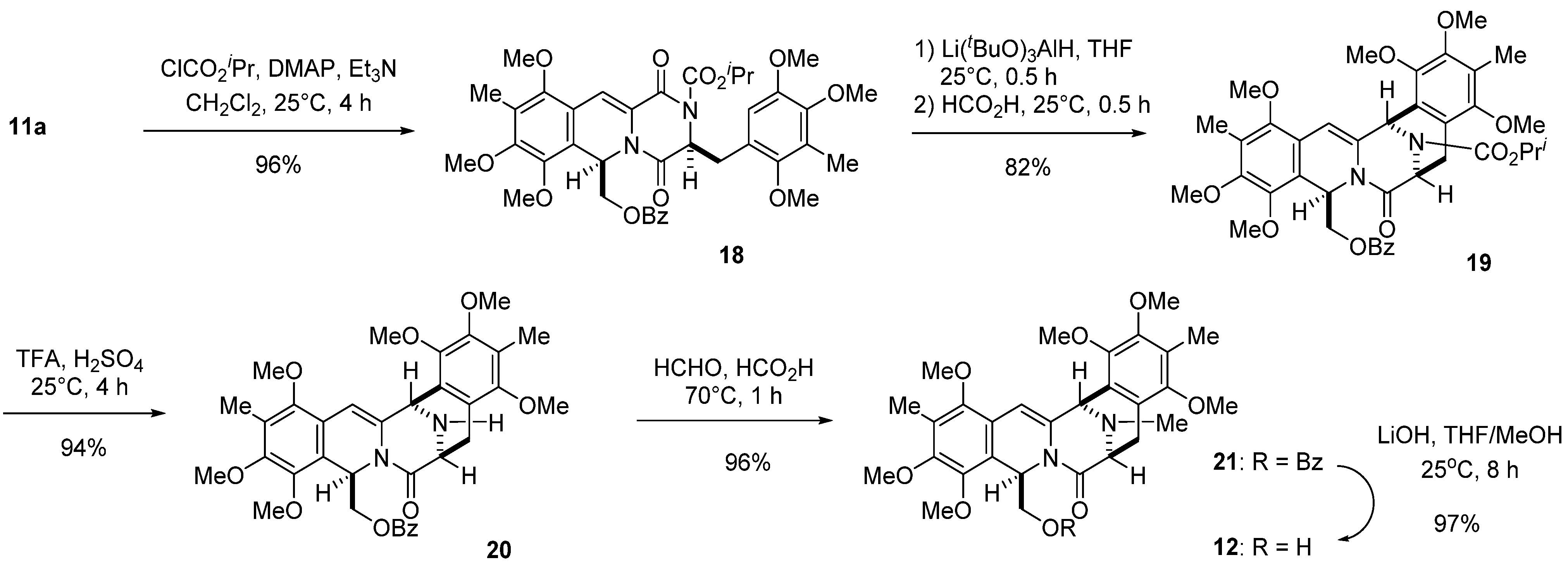
3.6. Isopropyl ((3S*,6R*)-7,8,10-Trimethoxy-9-methyl-1,4-dioxo-3-(2,4,5-trimethoxy-3-methylbenzyl)-1,3,4,6-tetrahydro-2H-pyrazino(1,2-b)isoquinolin-6-yl)methyl Benzoate (18)
A solution of 11a (16.47 g, 25 mmol), TEA (28.10 mL, 200 mmol), and DMAP (12.21 g, 100 mmol) in dichloromethane (400 mL) was cooled with ice water, and isopropyl chloroformate (40.08 mL, 350 mmol) was added dropwise over 30 min. The reaction mixture was stirred at 25 °C for 4 h. The organic layer was diluted with dichloromethane (500 mL), washed with 1 M aqueous HCl (500 mL ×2) and then water (500 mL), dried, and concentrated in vacuo to give a residue. The residue was subjected to column chromatography with ethyl acetate–hexane (2:5) to give 18 (17.5 g, 96%) as a yellow amorphous powder.
Compound 18: 1H-NMR (400 MHz, CDCl3) δ: 8.00 (2H, m, Ar-H), 7.48 (1H, m, Ar-H), 7.34 (2H, m, Ar-H), 7.19 (1H, s, C11-H), 6.53 (1H, t, J = 3.4 Hz, C6-H), 5.24 (1H, t, J = 6.1 Hz, C3-H), 4.96 (1H, sept, J = 6.3 Hz, CO2CH(CH3)2), 4.65 (1H, dd, J = 11.5, 3.4 Hz, C12-H), 4.36 (1H, dd, J = 11.5, 3.4 Hz, C12-H), 3.92 (3H, s, C7-OMe), 3.80 (3H, s, C4′-OMe or C5′-OMe), 3.78 (3H, s, C8-OMe), 3.63 (3H, s, C4′-OMe or C5′-OMe), 3.59 (3H, s, C2′-OMe), 3.44 (3H, s, C10-OMe), 3.34 (1H, dd, J = 13.7, 6.1 Hz, C3a-H), 3.17 (1H, dd, J = 13.7, 6.1 Hz, C3a-H), 2.10 (3H, s, C9-Me), 2.09 (3H, s, C3′-Me), 1.28 (3H, d, J = 6.3 Hz, CO2CH(CH3)2), 1.20 (3H, d, J = 6.3 Hz, CO2CH(CH3)2). 13C-NMR (100 MHz, CDCl3) δ: 165.7 (s, OCOPh), 163.4 (s, C4), 157.6 (s, C1), 153.4 (s, C8), 151.7 (s, CO2CH(CH3)2), 151.6 (s, C10), 151.4 (s, C2′), 149.0 (s, C4′ or C5′), 147.4 (s, C4′ or C5′), 145.8 (s, C7), 133.0 (d, Ph), 129.7 (d, Ph × 2), 129.6 (s, Ph), 128.3 (d, Ph × 2), 126.8 (s, C11a or C10a), 126.2 (s, C9), 125.6 (s, C3′), 122.5 (s, C1′), 120.0 (s, C6a), 119.3 (s, C11a or C12), 112.0 (d, C6′), 111.6 (d, C11), 71.8 (d, CO2CH(CH3)2), 66.3 (t, C12), 61.8 (q, C10-OCH3), 60.6 (q, C2′-OCH3), 60.5 (q, C7-OCH3), 60.0 (q, C4′-OCH3 or C5′-OCH3), 60.0 (q, C8-OCH3), 59.4 (d, C3), 55.9 (q, C4′-OCH3 or C5′-OCH3), 49.9 (d, C6), 35.8 (t, C3a), 21.6 (q, CO2CH(CH3)2), 21.5 (q, CO2CH(CH3)2), 9.7 (q, C3′-CH3), 9.2 (q, C9-CH3). FT-IR (KBr) cm−1: 2938, 1722, 1684, 1468, 1418, 1391, 1375, 1344, 1271, 1252, 1107, 1092, 1072, 712. EI-MS m/z (%): 732 (M+, 12), 597 (100), 569 (19), 555 (6), 511 (8), 483 (21), 415 (9), 260 (13), 232 (19), 195 (13). HR-EI-MS: calcd for C39H44N2O12, 732.2894, found: 732.2889.
3.7. Isopropyl (6S*,9R*,15R*)-9-((benzoyloxy)methyl)-1,2,4,10,11,13-hexamethoxy-3,12-dimethyl-7-oxo-6,7,9,15-tetrahydro-5H-6,15-epiminobenzo(4,5)azocino(1,2-b)isoquinoline-16-carboxylate (19)
A stirred solution of 18 (402.3 mg, 0.55 mmol) in THF (36 mL) was cooled with ice water and lithium tri-tert-butoxyaluminohydride (1.12 g, 4.4 mmol) was added over 10 min. After continued stirring at 25 °C for 30 min, anhydrous Na2SO4 (2 g) was added and the reaction mixture was quenched with water. The reaction mixture was filtered through Celite pad and then, the filtrate was diluted with brine (200 mL) and extracted with CHCl3 (3 × 200 mL). The combined extracts were washed with brine (200 mL), dried, and concentrated in vacuo to give a residue, which was used in the next step without further purification. A solution of the residue as above in formic acid (36 mL) was stirred at 25 °C for 30 min. The reaction mixture was concentrated in vacuo, and the residue was diluted with saturated aqueous NaHCO3 solution (80 mL) and extracted with CHCl3 (3 × 80 mL). The combined extracts were washed with brine (80 mL), dried, and concentrated in vacuo to give a residue. The residue was subjected to column chromatography with ethyl acetate–hexane (1:3) to give 19 (322.7 mg, 82%) as a pale yellow amorphous powder.
Compound 19: 1H-NMR (400 MHz, DMSO, 140 °C) δ: 7.58 (2H, m, Ar-H), 7.54 (1H, m, Ar-H), 7.41 (2H, m, Ar-H), 6.26 (1H, s, C14-H), 6.22 (1H, dd, J = 8.1, 4.2 Hz, C9-H), 5.93 (1H, d, J = 1.5, C15-H), 4.98 (1H, m, 1.5 Hz, C6-H), 4.86 (1H, sept, J = 6.2 Hz, CO2CH(CH3)), 3.92 (1H, dd, J = 11.5, 4.2 Hz, C16-H), 3.87 (3H, s, C2-OMe or C11-OMe), 3.87 (3H, s, C10-OMe), 3.85 (1H, dd, J = 11.5, 8.1 Hz, C16-H), 3.75 (3H, s, C4-OMe or C13-OMe), 3.73 (3H, s, C2-OMe or C11-OMe), 3.67 (3H, s, C4-OMe or C13-OMe), 3.51 (3H, s, C1-OMe), 3.09 (1H, br d, J =17.6 Hz, C5-H), 3.04 (1H, dd, J =17.6, 3.9 Hz, C5-H), 2.15 (3H, s, C3-Me or C12-Me), 1.98 (3H, s, C3-Me or C12-Me), 1.23 (3H, d, J = 6.2 Hz, CO2CH(CH3)2), 1.21 (3H, d, J = 6.2 Hz, CO2CH(CH3)2). 13C-NMR (100 MHz, DMSO, 140 °C) δ: 165.4 (s, C7), 164.4 (s, OCOPh), 152.1 (s, CO2CH(CH3)2), 151.7 (s, C1), 150.2 (s, C2 or C11), 149.2 (s, C4 and C13), 145.0 (s, C2 or C11), 144.7 (s, C10), 132.3 (s, C14a), 131.9 (d, Ph), 128.7 (s, Ph), 128.3 (d, Ph × 2), 127.5 (d, Ph × 2), 124.5 (s, C3 or C12), 124.0 (s, C3 or C12), 123.7 (s, C4a), 119.9 (s, C15a), 118.8 (s, C13a), 118.2 (s, C9a), 100.0 (d, C14), 68.8 (d, CO2CH(CH3)2), 62.6 (t, C16), 60.3 (q, C4-OCH3 or C13-OCH3), 59.8 (q, C2-OCH3 or C10-OCH3 or C11-OCH3), 59.2 (q, C2-OCH3 or C10-OCH3 or C11-OCH3), 59.1 (q, q, C2-OCH3 or C10-OCH3 or C11-OCH3), 58.9 (q, C4-OCH3 or C13-OCH3), 58.6 (q, C1-OCH3), 52.5 (d, C6), 48.9 (d, C15), 45.5 (d, C9), 26.9 (t, C5), 20.9 (q, CO2CH(CH3)2), 20.9 (q, CO2CH(CH3)2), 8.24 (q, C3-CH3 or C12-CH3), 8.18 (q, C3-CH3 or C12-CH3). FT-IR (KBr) cm−1: 1717, 1707, 1686, 1647, 1466, 1414, 1362, 1344, 1298, 1269, 1109, 1070, 1007, 964, 712. EI-MS m/z (%): 716 (M+, 22), 581 (100), 553 (67), 234 (21). HR-EI-MS: calcd for C39H44N2O11, 716.2945, found: 716.2942.
3.8. ((6S*,9R*,15R*)-1,2,4,10,11,13-hexamethoxy-3,12-dimethyl-7-oxo-6,7,9,15-tetrahydro-5H-6,15-epiminobenzo(4,5)azocino(1,2-b)isoquinolin-9-yl)methyl Benzoate (20)
Concentrated H2SO4 (1.7 mL) was added to a stirred solution of 19 (322.7 mg, 0.45 mmol) in TFA (34 mL) at 0 °C over 5 min, and the reaction mixture was stirred at 25 °C for 4 h. The reaction mixture was poured into water (40 mL) at 0 °C, basified with concentrated NH4OH, and then extracted with CHCl3 (3 × 100 mL). The combined extracts were washed brine (100 mL), dried, and concentrated in vacuo to give a residue. The residue was subjected to column chromatography with ethyl acetate–hexane (1:3) to give 20 (267.6 mg, 94%) as a pale yellow amorphous powder.
Compound 20: 1H-NMR (400 MHz, CDCl3) δ: 7.70 (2H, m, Ar-H), 7.49 (1H, m, Ar-H), 7.38 (2H, m, Ar-H), 6.41 (1H, dd, J = 8.1, 5.2 Hz, C9-H), 6.26 (1H, s, C14-H), 4.99 (1H, s, C15-H), 4.12 (1H, br d, J = 6.3 Hz, C6-H), 3.95~3.89 (2H, overlapped, C16-H), 3.90 (3H, s, C10-OMe), 3.87 (3H, s, C1-OMe), 3.76 (3H, s, C13-OMe), 3.76 (3H, s, C11-OMe), 3.67 (3H, s, C2-OMe), 3.44 (3H, s, C4-OMe), 3.19 (1H, dd, J = 17.0, 1.3 Hz, C5-H), 3.07 (1H, dd, J = 17.0, 6.3 Hz, C5-H), 2.19 (3H, s, C12-Me), 1.88 (3H, s, C3-Me). 13C-NMR (100 MHz, CDCl3) δ: 168.5 (s, C7), 166.0 (s, OCOPh), 152.5 (s, C4), 150.8 (s, C11), 149.9 (s, C2), 149.8 (s, C13), 146.3 (s, C1), 146.0 (s, C10), 136.1 (s, C13a or C14a), 132.5 (d, Ph), 129.6 (d, Ph), 129.4 (s, Ph), 128.3 (d, Ph), 125.7 (s, C12), 125.3 (s, C15a), 125.0 (s, C3), 121.5 (s, C4a), 120.4 (s, C13a or C14a), 119.5 (s, C9a), 99.8 (d, C14), 63.4 (t, C16), 61.4 (q, C13-OCH3), 60.8 (q, C10-OCH3), 60.2 (q, C1-OCH3), 60.1 (q, C11-OCH3), 59.9 (q, C2-OCH3), 59.3 (q, C4-OCH3), 53.9 (d, C6), 50.1 (d, C15), 45.7 (d, C9), 29.0 (t, C5), 9.3 (q, C12-CH3), 9.1 (q, C3-CH3). FT-IR (KBr) cm−1: 2938, 1722, 1680, 1638, 1466, 1412, 1362, 1271, 1248, 1115, 1070, 1009, 964, 712. EI-MS m/z (%): 630 (M+, 22), 495 (48), 467 (100), 234 (39), 204 (18). HR-EI-MS: calcd for C35H38N2O9, 630.2577, found: 630.2581.
3.9. ((6S*,9R*,15R*)-1,2,4,10,11,13-hexamethoxy-3,12,16-trimethyl-7-oxo-6,7,9,15-tetrahydro-5H-6,15-epiminobenzo(4,5)azocino(1,2-b)isoquinolin-9-yl)methyl Benzoate (21)
A 37% aqueous solution of formaldehyde (6 mL) was added to a stirred solution of 20 (251.2 mg, 0.4 mmol) in formic acid (7.0 mL) at 60 °C, and the reaction mixture was heated at 70 °C for 1 h. The reaction mixture was diluted with 5% aqueous NaHCO3 solution (80 mL) and extracted with CHCl3 (3 × 80 mL). The combined extracts were washed brine (80 mL), dried, and concentrated in vacuo to give a residue. The residue was subjected to column chromatography with ethyl acetate–hexane (1:1) to give 21 (247.3 mg, 96%) as a colorless amorphous powder.
Compound 21: 1H-NMR (400 MHz, CDCl3) δ: 7.71 (2H, m, Ar-H), 7.49 (1H, m, Ar-H), 7.38 (2H, m, Ar-H), 6.43 (1H, t, J = 6.4 Hz, C9-H), 6.30 (1H, s, C14-H), 4.63 (1H, s, C15-H), 3.92 (3H, s, C10-OMe), 3.91 (2H, d, J = 6.4 Hz, C17-H), 3.87 (3H, s, C1-OMe), 3.76 (6H, s, C11-OMe and C13-OMe), 3.68 (1H, overlapped, C6-H), 3.67 (3H, s, C2-OMe), 3.43 (3H, s, C4-OMe), 3.15 (1H, dd, J = 17.6, 4.1 Hz, C5-H), 3.11 (1H, br d, J = 17.6 Hz, C5-H), 2.55 (3H, s, N-Me), 2.20 (3H, s, C12-Me), 1.89 (3H, s, C3-Me). 13C-NMR (100 MHz, CDCl3) δ: 168.1 (s, C7), 166.0 (s, OCOPh), 152.4 (s, C4), 150.7 (s, C11), 149.8 (s, C2), 149.5 (s, C13), 146.1 (s, C1), 145.9 (s, C10), 132.7 (s, C14a), 132.5 (d, Ph), 129.6 (d, Ph × 2), 129.3 (s, Ph), 128.3 (d, Ph × 2), 126.0 (s, C15a), 125.7 (s, C12), 124.7 (s, C3), 121.1 (s, C4a), 119.9 (s, C13a), 119.6 (s, C9a), 102.7 (d, C14), 63.6 (t, C17), 61.3 (q, C11-OCH3 or C13-OCH3), 60.7 (q, C10-OCH3), 60.5 (d, C6), 60.1 (q, C1-OCH3), 60.0 (q, C11-OCH3 or C13-OCH3), 59.8 (q, C2-OCH3), 59.2 (q, C4-OCH3), 56.8 (d, C15), 45.5 (d, C9), 41.6 (q, N-CH3), 29.2 (t, C5), 9.2 (q, C12-CH3), 9.0 (q, C3-CH3). FT-IR (KBr) cm−1: 1722, 1678, 1638, 1464, 1412, 1358, 1341, 1271, 1126, 1113, 1099, 1067, 1007, 712. EI-MS m/z (%): 644 (M+, 17), 509 (20), 481 (100), 248 (37), 218 (15). HR-EI-MS: calcd for C36H40N2O9, 644.2734, found: 644.2733.
3.10. (6S*,9R*,15R*)-9-(hydroxymethyl)-1,2,4,10,11,13-hexamethoxy-3,12,16-trimethyl-5,6,9,15-tetrahydro-7H-6,15-epiminobenzo(4,5)azocino(1,2-b)isoquinolin-7-one (12)
A 10 M aqueous solution of lithium hydroxide monohydrate (77 μL, 0.77 mmol) was added to a stirred solution of 21 (226.3 mg, 0.35 mmol) in THF (2.0 mL) and MeOH (0.7 mL), and stirring was continued at 25 °C for 8 h. The reaction mixture was diluted with water (80 mL), and the mixture was extracted with CHCl3 (3 × 80 mL). The combined extracts were washed with brine (80 mL), dried, and concentrated in vacuo to give a residue. The residue was subjected to column chromatography with ethyl acetate–hexane (2:1) to give 12 (184.1 mg, 97%) as a colorless amorphous powder.
Compound 12: 1H-NMR (400 MHz, CDCl3) δ: 6.27 (1H, s, C14-H), 6.08 (1H, dd, J = 7.9, 5.1 Hz, C9-H), 4.70 (1H, brs, C15-H), 3.88 (3H, s, OMe), 3.86 (3H, s, OMe), 3.78 (3H, s, OMe), 3.77 (3H, s, OMe), 3.75 (3H, s, OMe) 3.72 (1H, br t, C6-H), 3.66 (3H, s, OMe), 3.32 (1H, dt, J = 11.5, 5.1 Hz, C17-H), 3.22 (1H, dt, J = 11.5, 7.9 Hz, C17-H), 3.21 (2H, d, J = 4.4 Hz, C5-H), 2.57 (3H, s, N-Me), 2.19 (3H, s, C12-Me), 2.18 (3H, s, C3-Me), 1.37 (1H, t, J = 6.1 Hz, -OH). 13C-NMR (100 MHz, CDCl3) δ: 169.1 (C7), 152.6 (C4), 150.7 (C11), 150.1 (C2), 149.7 (C13), 146.3 (C1), 145.8 (C10), 132.8 (C14a), 126.0 (C15a), 125.4 (C12), 125.1 (C3), 121.1 (C4a), 120.6 (C9a), 119.5 (C13a), 102.9 (14), 64.6 (C17), 61.3 (-OCH3), 60.6 (-OCH3), 60.6 (C6), 60.1 (-OCH3), 60.1 (-OCH3), 60.0 (-OCH3), 59.8 (-OCH3), 56.5 (C15), 49.1 (C9), 41.6 (N-CH3), 29.3 (C5), 9.4 (-CH3), 9.2 (-CH3). FT-IR (KBr) cm−1: 3468, 2940, 1672, 1636, 1466, 1412, 1341, 1248, 1113, 1065, 1007, 964. EI-MS m/z (%): 540 (M+, 9), 509 (27), 481 (100), 248 (51). HR-EI-MS: calcd for C29H36N2O8, 540.2472, found: 540.2473.
3.11. ((6S*,9R*,15R*)-1,2,4,10,11,13-hexamethoxy-3,12,16-trimethyl-7-oxo-6,7,9,15-tetrahydro-5H-6,15-epiminobenzo(4,5)azocino(1,2-b)isoquinolin-9-yl)methyl Benzoate (22)
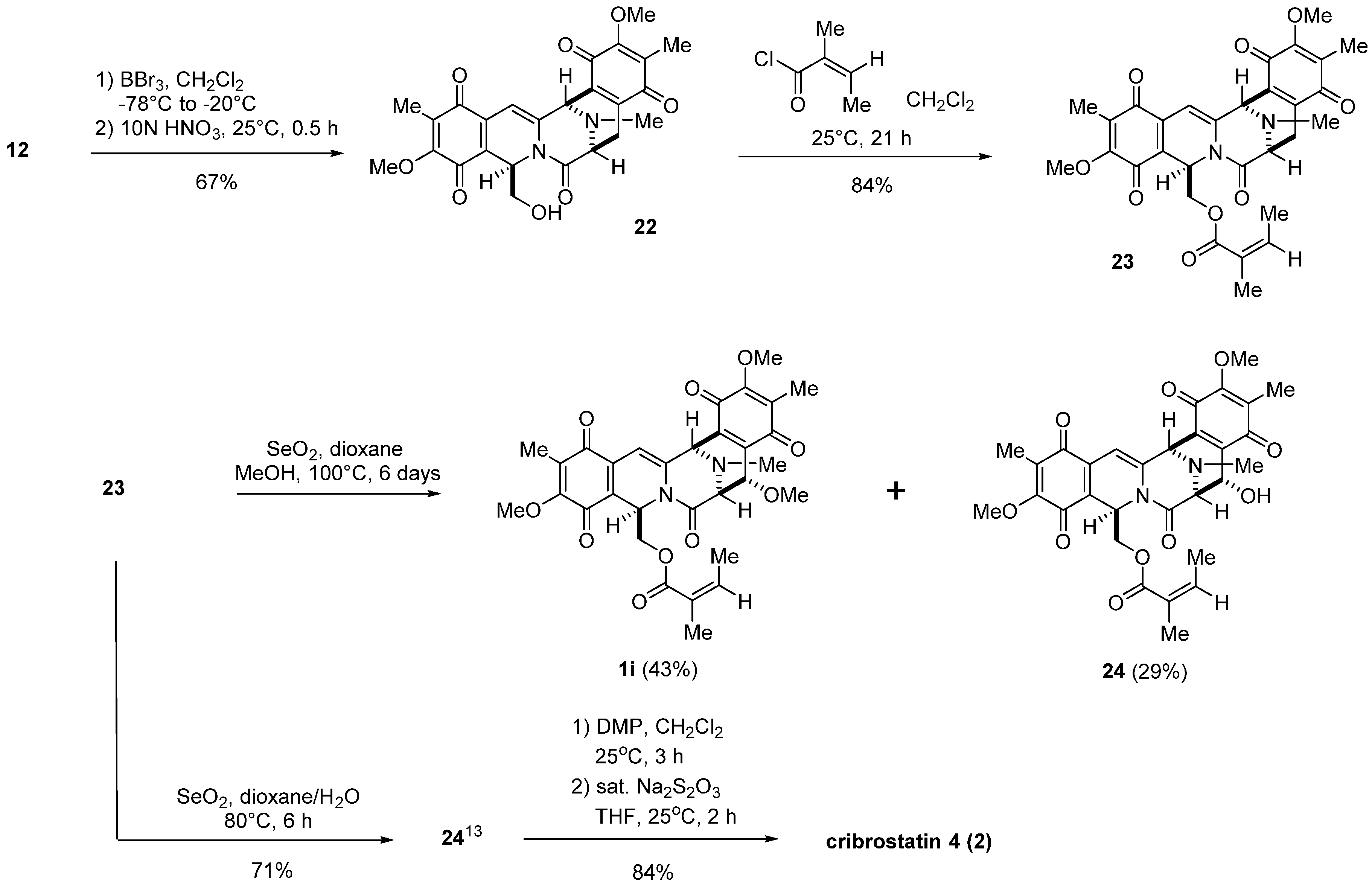
To a stirred solution of 12 (216.0 mg, 0.4 mmol) in CH2Cl2 (24 mL) at −78 °C was added a CH2Cl2 solution of BBr3 (1.0 M, 2.40 mL, 2.4 mmol) over 5 min. Stirring was continued at the same temperature for 1 h, and then at −20 °C for 14.5 h. The reaction mixture was diluted with water (200 mL) and extracted with 5% MeOH in CHCl3 (4 × 200 mL). The combined extracts were washed with 5% NaHCO3 solution (200 mL), dried, and concentrated in vacuo to give a residue. A solution of the above residue in 10 N HNO3 (5.0 mL) was stirred at 25 °C for 30 min. The reaction mixture was diluted with water (150 mL) and extracted with ethyl acetate (3 × 200 mL). The combined extracts were washed with brine (200 mL), dried, and concentrated in vacuo. The residue was subjected to purification by silica gel chromatography with ethyl acetate to give 22 (130.0 mg, 67%) as a dark purple amorphous powder.
Compound 22: 1H-NMR (500 MHz, CDCl3) δ: 6.26 (1H, s, 14-H), 5.96 (1H, dd, J = 7.1, 4.5 Hz, 9-H), 4.55 (1H, s, 15-H), 4.02 (3H, s, OCH3), 3.97 (3H, s, OCH3), 3.74 (1H, dt, J = 6.5, 1.5 Hz, 6-H), 3.50 (1H, dd, J = 11.4, 4.5 Hz, 17-H), 3.36 (1H, dd, J = 11.4, 7.1 Hz, 17-H), 2.96 (1H, dd, J = 19.8, 6.5 Hz, 5-Hα), 2.89 (1H, dd, J = 19.8, 1.5 Hz, C5-Hβ), 2.51 (3H, s, N-CH3), 1.96 (3H, s, 3-CH3 or 12-CH3), 1.94 (3H, s, 3-CH3 or 12-CH3), 1.62 (1H, br s, OH). 13C-NMR (125 MHz, CDCl3) δ: 186.6 (s, C-4), 185.0 (s, C-13), 180.5 (s, C-10 and C-1), 168.0 (s, C-7), 156.0 (s, C-11), 155.4 (s, C-2), 140.7 (s, C-14a), 140.0 (s, C-4a), 136.5 (s, C-15a), 134.5 (s, C-13a), 129.1 (s, C-3), 127.6 (s, C-12), 125.0 (s, C-9a), 101.8 (d, C-14), 62.9 (t, C-17), 61.1 (q, OCH3), 61.0 (q, OCH3), 59.6 (d, C-6), 54.4 (d, C-15), 48.4 (d, C-9), 41.2 (q, N-CH3), 28.7 (t, C-5), 8.8 (q, Ar-CH3), 8.7 (q, Ar-CH3). FT-IR (KBr) cm−1: 3347, 2951, 2855, 1654, 1616, 1568, 1450, 1373, 1310. LR-MS (FAB+): 481 [M + H]+. HR-MS (FAB+): calcd for C25H25N2O8, 481.1611, found: 481.1623.
3.12. ((6S*,9R*,15R*)-2,11-dimethoxy-3,12,16-trimethyl-1,4,7,10,13-pentaoxo-1,5,6,7,9,10,13,15-octa-hydro-4H-6,15-epiminobenzo(4,5)azocino(1,2-b)isoquinolin-9-yl)methyl (Z)-2-methylbut-2-enoate (23)
A solution of angelic acid (601.0 mg, 6.0 mmol) in ether (30 mL) was cooled with ice water, and a solution of oxalyl chloride (0.5 mL, 5.9 mmol) in DMF (46.0 μL, 592 mmol) was added dropwise over 5 min. The resulting solution was stirred at 25 °C for 2 h. Then, a solution of 22 (142.0 mg, 0.3 mmol) in CH2Cl2 (15 mL) was added over 5 min. The reaction mixture was concentrated to approximately 3.0 mL with a stream of argon, and CH2Cl2 (8.0 mL) was added. The resulting mixture was stirred at 25 °C for 21 h. The reaction mixture was directly purified by silica gel chromatography with ethyl acetate–hexane (2:1) to afford 23 (139.0 mg, 84%) as a dark purple film.
Compound 23: 1H-NMR (500 MHz, CDCl3) δ: 6.24 (1H, s, 14-H), 6.12 (1H, dd, J = 5.7, 2.9 Hz, 9-H), 5.92 (1H, qq, J = 7.4, 1.4 Hz, 21-H), 4.50 (1H, br s, 15-H), 4.21 (1H, dd, J = 11.9, 5.7 Hz, 17-H), 4.05 (3H, s, C11-OCH3), 4.01 (3H, s, 2-OCH3), 4.01 (1H, dd, J = 11.9, 2.9 Hz, 17-H), 3.72 (1H, dt, J = 6.8, 1.4 Hz, 6-H), 2.95 (1H, dd, J = 19.8, 6.8 Hz, 5-Hβ), 2.84 (1H, dd, J = 19.8, 1.4 Hz, 5-H), 2.47 (3H, s, N-CH3), 1.96 (3H, s, 12-CH3), 1.92 (3H, s, 3-CH3), 1.75 (3H, dq, J = 7.4, 1.4 Hz, 21-CH3), 1.57 (1H, quint, J = 1.4 Hz, 20-CH3). 13C-NMR (125 MHz, CDCl3) δ: 186.5 (s, C-4), 184.9 (s, C-13), 180.5 (s, C-1), 180.1 (s, C-10), 167.1 (s, C-7 and C-19), 156.2 (s, C-11), 155.2 (s, C-2), 140.6 (s, C-14a), 139.8 (s, C-4a), 139.3 (d, C-21), 136.2 (s, C-15a), 134.6 (s, C-13a), 128.5 (s, C-3), 127.3 (s, C-12), 126.8 (s, C-20), 124.2 (s, C-9a), 101.3 (d, C-14), 62.4 (t, C-17), 61.1 (q, 11-OCH3), 61.0 (q, 2-OCH3), 59.5 (d, C-6), 54.3 (d, C-15), 47.1 (d, C-9), 41.1 (q, N-CH3), 28.3 (t, C-5), 20.2 (q, C-23), 15.5 (q, C-22), 8.7 (q, 3-CH3), 8.6 (q, 12-CH3). FT-IR (KBr) cm−1: 2949, 1653, 1616, 1570, 1458, 1309, 1228, 1153. EI-MS m/z (%): 562 (M+, 5), 421 (100), 218 (40). HR-EI-MS: calcd for C30H30N2O9, 562.1951, found: 562.1952.
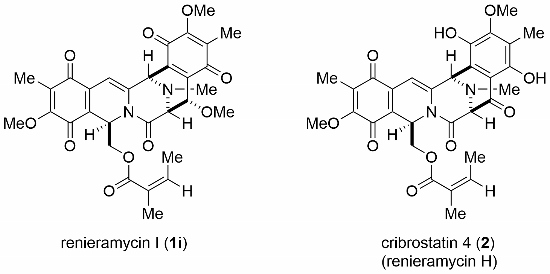
3.13. Renieramycin I (1i)
A suspension of 23 (15.0 mg, 0.027 mmol) and SeO2 (29.6 mg, 0.27 mmol) in dioxane (3.0 mL) and MeOH (1.0 mL) was heated at 100 °C for 6 days. The reaction mixture was filtered and the filter cake was washed with ethyl acetate. The combined filtrates were concentrated in vacuo to give a residue. Flash column chromatography on silica gel with ethyl acetate–hexane (2:3) afforded 1i (6.8 mg, 43%) as a dark red film and 24 (4.4 mg, 29%).
Renieramycin I (1i): 1H-NMR (400 MHz, CDCl3) δ: 6.26 (1H, s, 4-H), 6.07 (1H, dd, J = 5.5, 2.7 Hz, 1-H), 5.92 (1H, qq, J = 7.3, 1.4 Hz, 26-H), 4.54 (1H, d, J = 0.9 Hz, 11-H), 4.34 (1H, d, J = 1.4 Hz, 14-H), 4.16 (1H, dd, J = 12.1, 5.5 Hz, 22-H), 4.06 (3H, s, 7-OCH3), 4.02 (1H, dd, J = 12.1, 2.7 Hz, 22-H), 3.98 (3H, s, 17-OCH3), 3.74 (1H, br t, J = 1.4 Hz, 13-H), 3.62 (3H, s, 14-OCH3), 2.55 (3H, s, N-CH3), 1.96 (3H, s, 6-CH3), 1.94 (3H, s, 16-CH3), 1.73 (3H, dq, J = 7.3, 1.4 Hz, 26-CH3), 1.55 (3H, quint, J = 1.4 Hz, 25-CH3). 13C-NMR (100 MHz, CDCl3) δ: 185.8 (s, C-15), 184.7 (s, C-5), 180.9 (s, C-18), 180.1 (s, C-8), 167.1 (s, C-24), 164.1 (s, C-21), 156.1 (s, C-7), 155.2 (s, C-17), 139.2 (d, C-26), 138.6 (s, C-3), 137.4 (s, C-19), 137.0 (s, C-20), 134.2 (s, C-10), 129.3 (s, C-16), 127.4 (s, C-6), 126.7 (s, C-25), 124.9 (s, C-9), 102.2 (d, C-4), 73.4 (d, C-14), 64.7 (d, C-13), 62.6 (t, C-22), 61.2 (q, 7-OCH3), 60.9 (q, 17-OCH3), 59.4 (q, 14-OCH3), 54.5 (d, C-11), 47.3 (d, C-1), 41.7 (q, N-CH3), 20.2 (q, 25-CH3), 15.5 (q, 26-CH3), 8.9 (q, Ar-CH3), 8.7 (q, Ar-CH3). FT-IR (KBr) cm−1: 3429, 2949, 1717, 1684, 1655, 1614, 1570, 1454, 1342, 1306, 1233, 1209, 1153, 1096. EI-MS m/z (%): 594 ([M + 2H]+, 0.7), 593 ([M + H]+, 2), 592 (M+, 6), 479 (19), 452 (26), 451 (100), 421 (20), 248 (25), 218 (11). HR-EI-MS: calcd for C30H30N2O10, 592.2057, found: 592.2056.
3.14. ((6S*,9R*,15R*)-5-hydroxy-2,11-dimethoxy-3,12,16-trimethyl-1,4,7,10,13-pentaoxo-1,5,6,7,9,10,13,15-octahydro-4H-6,15-epiminobenzo(4,5)azocino(1,2-b)isoquinolin-9-yl)methyl (Z)-2-methylbut-2-enoate (24)
1H-NMR (500 MHz, CDCl3) δ: 6.28 (1H, s, 4-H), 6.09 (1H, dd, J = 6.0, 2.9 Hz, 1-H), 5.93 (1H, qq, J = 7.3, 1.5 Hz, 26-H), 4.86 (1H, br d, J = 7.0 Hz, 14-H), 4.52 (1H, d, J = 1.1 Hz, 11-H), 4.19 (1H, dd, J = 12.0, 6.0 Hz, 22-H), 4.06 (3H, s, 7-OCH3), 4.01 (3H, s, 17-OCH3), 3.99 (1H, dd, J = 12.0, 2.9 Hz, 22-H), 3.75 (1H, dd, J = 1.7, 1.1 Hz, 13-H), 2.88 (1H, br d, J = 7.0 Hz, OH), 2.55 (3H, s, N-CH3), 1.96 (3H, s, 6-CH3), 1.93 (3H, s, 16-CH3), 1.75 (3H, dq, J = 7.3, 1.5 Hz, 26-CH3), 1.56 (3H, quint, J = 1.5 Hz, 25-CH3). 13C-NMR (125 MHz, CDCl3) δ: 186.7 (s, C-15), 184.7 (s, C-5), 181.0 (s, C-18), 180.0 (s, C-8), 167.1 (s, C-24), 163.9 (s, C-21), 156.1 (s, C-7), 155.4 (s, C-17), 139.3 (d, C-26), 138.4 (s, C-10 or C-20), 138.3 (s, C-10 or C-20), 136.9 (s, C-19), 134.3 (s, C-3), 128.8 (s, C-16), 127.4 (s, C-6), 126.7 (s, C-25), 124.8 (s, C-9), 102.2 (d, C-4), 67.1 (d, C-13), 64.8 (d, C-14), 62.3 (t, C-22), 61.1 (q, OCH3), 61.0 (q, OCH3), 54.7 (d, C-11), 47.2 (d, C-1), 41.5 (q, N-CH3), 20.2 (q, 25-CH3), 15.5 (q, 26-CH3), 8.7 (q, Ar-CH3), 8.7 (q, Ar-CH3). FT-IR (KBr) cm−1: 3446, 2930, 2857, 1654, 1616, 1570, 1456, 1307, 1233, 1211, 1153. EI-MS m/z (%): 578 (M+, 5), 437 (100), 421 (48), 218 (20). HR-EI-MS: calcd for C30H30N2O10, 578.1900, found: 578.1899.
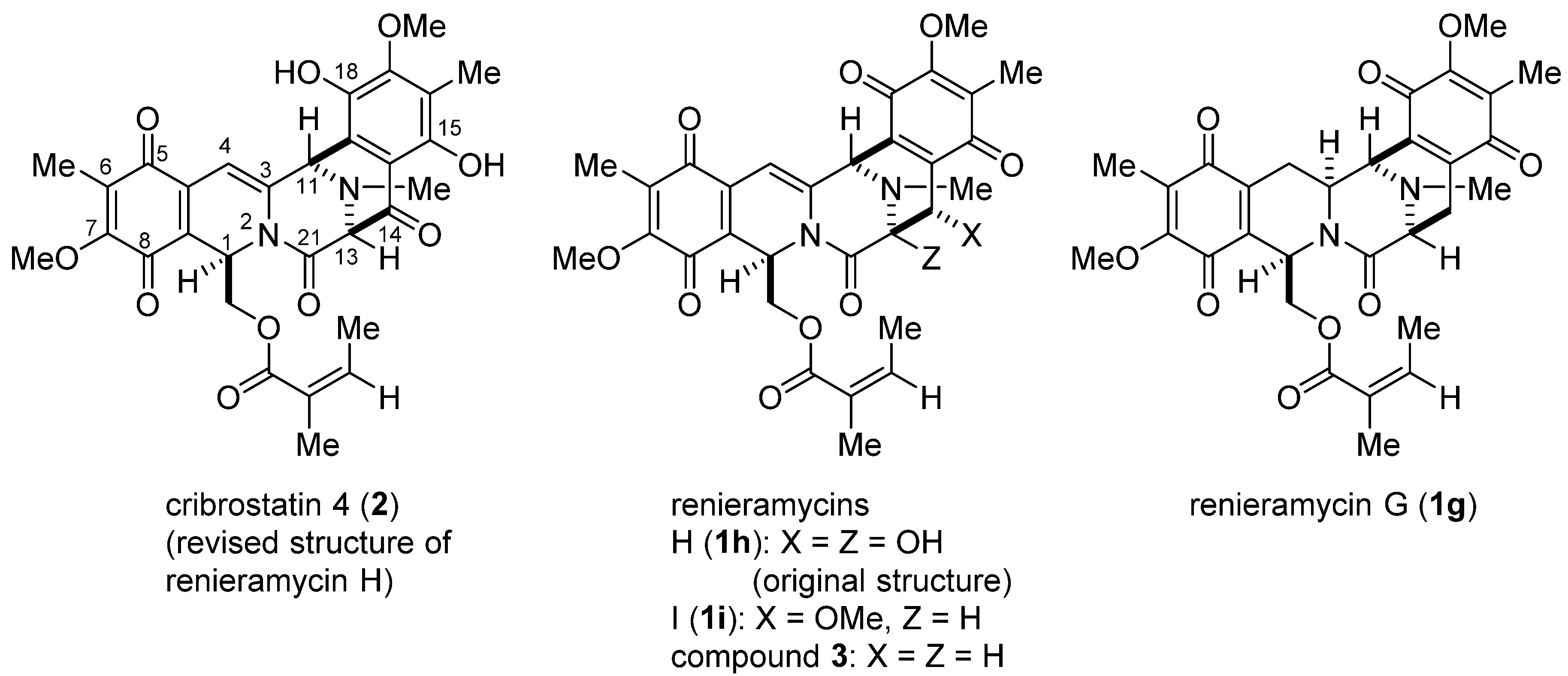
3.15. Cribrostatin 4 (2) via 24
A suspension of
23 (112.4 mg, 0.2 mmol) and SeO
2 (110.9 mg, 1.0 mmol) in dioxane (30 mL) and water (3.0 mL) was heated at 80 °C for 6 h. The reaction mixture was filtered and the filter cake was washed with ethyl acetate (200 mL). The combined filtrates were concentrated
in vacuo to give a residue (248.3 mg). Flash column chromatography on silica gel (70 g) with hexane–ethyl acetate (1:1) afforded
24 (72.5 mg, 63%) along with recovered
23 (16.2 mg, 14%). Dess-Martin periodinane (DMP, 445.3 mg, 1.05 mmol) was added to a stirred solution of
24 (57.8 mg, 0.1 mmol) in dichloromethane (15 mL), and the mixture was stirred at 25 °C for 3 h. The reaction mixture was diluted with THF (50 mL), saturated aqueous Na
2S
2O
3 solution (50 mL) was added, and the mixture was stirred at 25 °C for 2 h. The reaction mixture was diluted with water (100 mL) and extracted with ethyl acetate (100 mL × 3). The combined extracts were washed with brine (100 mL), dried, and concentrated
in vacuo. The residue (230 mg) was purified by silica gel (9 g) flash column chromatography with hexane–ethyl acetate (1:2) to give cribrostatin 4 (
2: 81.0 mg, 70.0% from
23) as a dark red film. 1H-, 13C NMR, and also IR spectral charts of synthetic renieramycin
I and cribrostatin
4 are available in the
supplementary information.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







