
Epipolythiodiketopiperazines from the Marine Derived Fungus Dichotomomyces cejpii with NF-κB Inhibitory Potential



Abstract
:1. Introduction
2. Results and Discussion
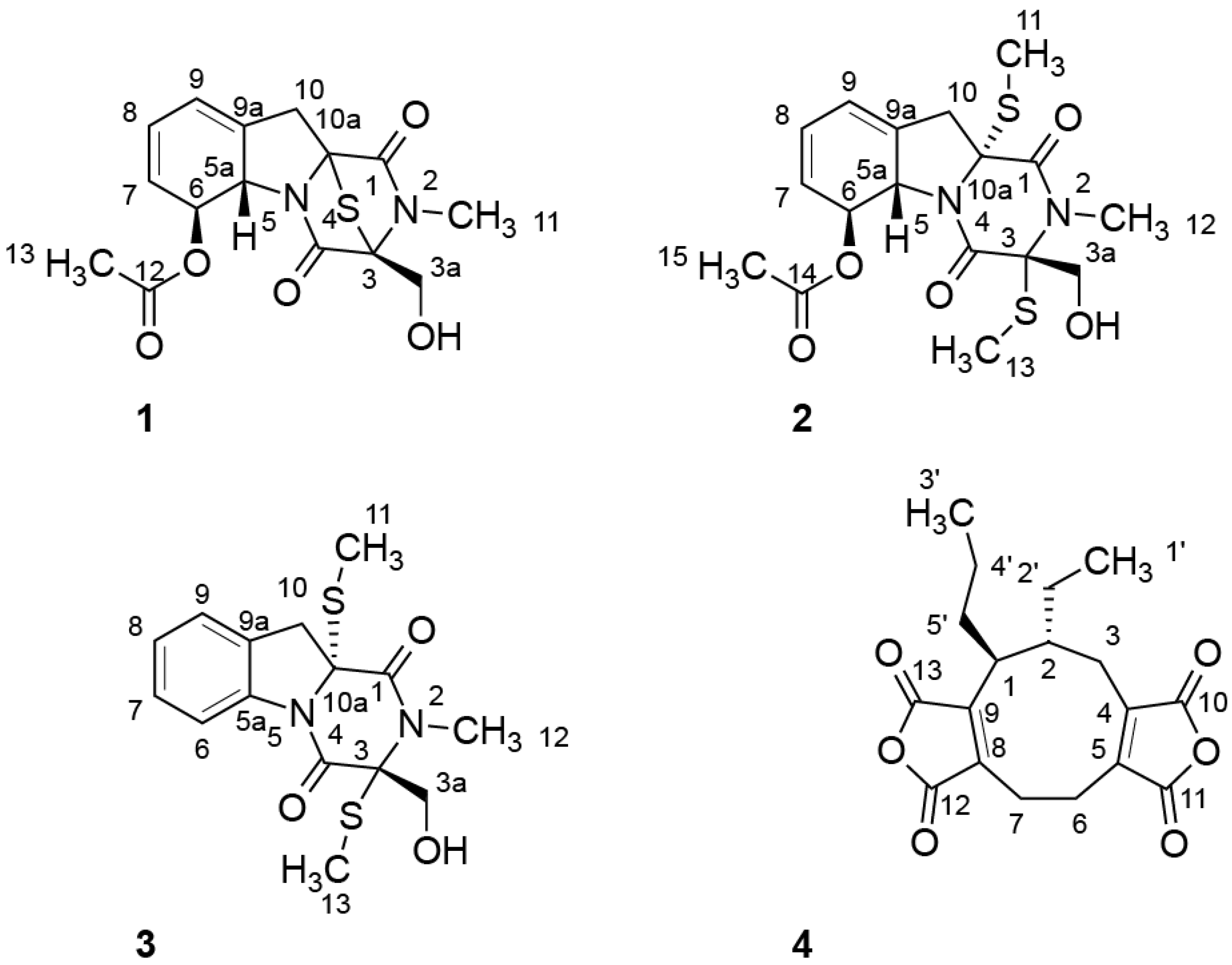
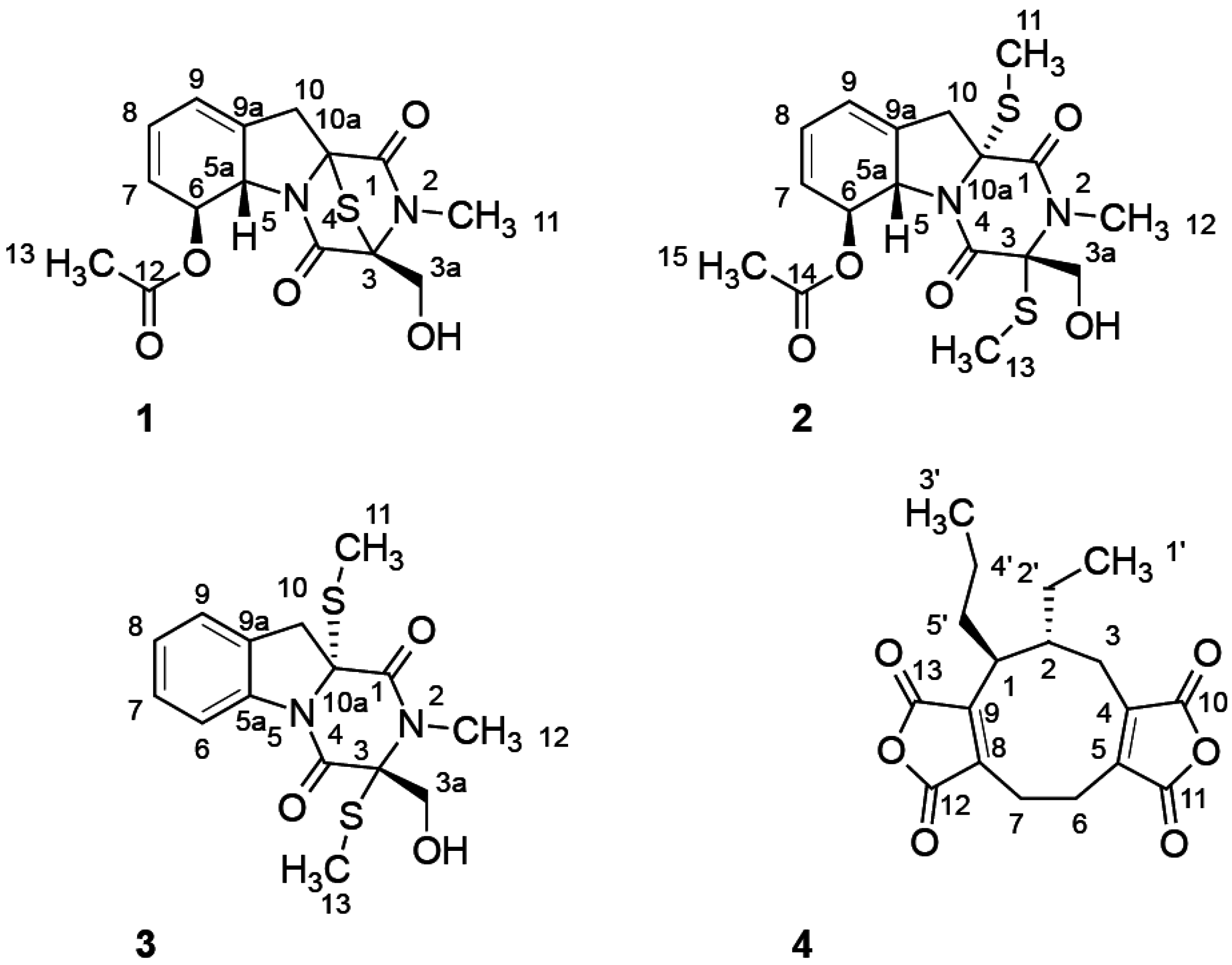
2.1. Structure Elucidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Compound 1 | Compound 2 | Compound 3 | |||
|---|---|---|---|---|---|---|
| δC/N, Type | δH (J in Hz) | δC/N, Type | δH (J in Hz) | δC/N, Type | δH (J in Hz) | |
| 1 | 174.2, C | - | 166.8, C | - | 166.3, C | - |
| 2 | N | - | N | - | N | - |
| 3 | 82.5, C | - | 74.5, C | - | 73.2, C | - |
| 3a | 58.0, CH2 | 3a′: 4.30, dd (5.9, 13.2) 3a″: 4.17, dd (5.9, 13.2) | 64.3, CH2 | 3a′: 4.20, dd (5.9, 11.3) 3a″: 3.72, dd (5.9, 11.3) | 64.8, CH2 | 3a′: 4.42, dd (4.0, 12.1) 3a″: 3.95, dd (4.0, 12.1) |
| 4 | 171.3, C | - | 164.9, C | - | 162.9, C | - |
| 5 | N | - | N | - | N | - |
| 5a | 61.6, CH | 4.57, br d (13.5) | 66.3, CH | 5.10, br d (13.5) | 142.4, C | - |
| 6 | 74.9, CH | 5.79, br d (13.5) | 75.7, CH | 6.17, br d (13.5) | 118.3, CH | 8.02, d (7.3) |
| 7 | 127.1, CH | 5.53, br d (9.9) | 128.2, CH | 5.57, br d (9.9) | 128.3, CH | 7.30, t (7.3) |
| 8 | 125.7, CH | 6.01, m | 126.2, CH | 6.01, m | 126.3, CH | 7.19, t (7.3) |
| 9 | 119.5, CH | 6.03, br s | 120.0, CH | 6.03, br s | 126.2, CH | 7.39, d (7.3) |
| 9a | 137.4, C | - | 135.8, C | - | 130.3, C | - |
| 10 | 29.2, CH2 | 10′: 3.45, d (18.3) 10″: 2.98, d (18.3) | 40.4, CH2 | 10′: 3.09, d (15.7) 10″: 2.79, d (15.7) | 40.0, CH2 | 10′: 3.59, d (16.8) 10″: 3.51, d (16.8) |
| 10a | 80.4, C | - | 73.6, C | - | 71.6, C | - |
| 11 | 27.8, CH3 | 2.98, s | 14.9, CH3 | 2.25, s | 14.4, CH3 | 2.22, s |
| 12 | 170.5, C | - | 28.7, CH3 | 3.03, s | 28.9, CH3 | 3.15, s |
| 13 | 21.2, CH3 | 2.06, s | 12.7, CH3 | 2.18, s | 13.5, CH3 | 2.32, s |
| 14 | 170.6, C | - | ||||
| 15 | 21.3, CH3 | 2.03, s | ||||
| 3a-OH | - | 4.74, br t (5.9) | - | 4.50, br t (5.9) | 4.62, br t (4.0) | |

2.2. Bioactivity
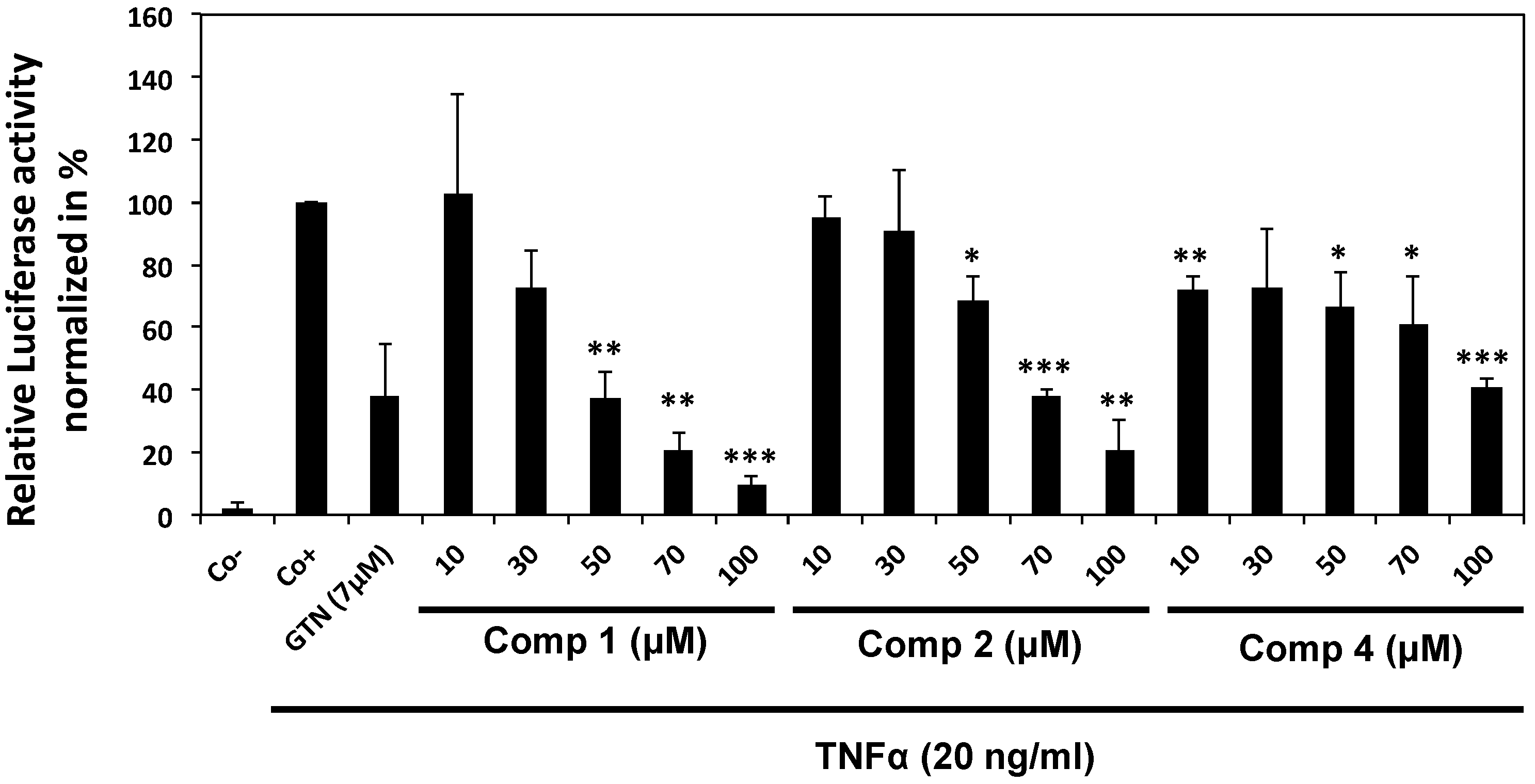
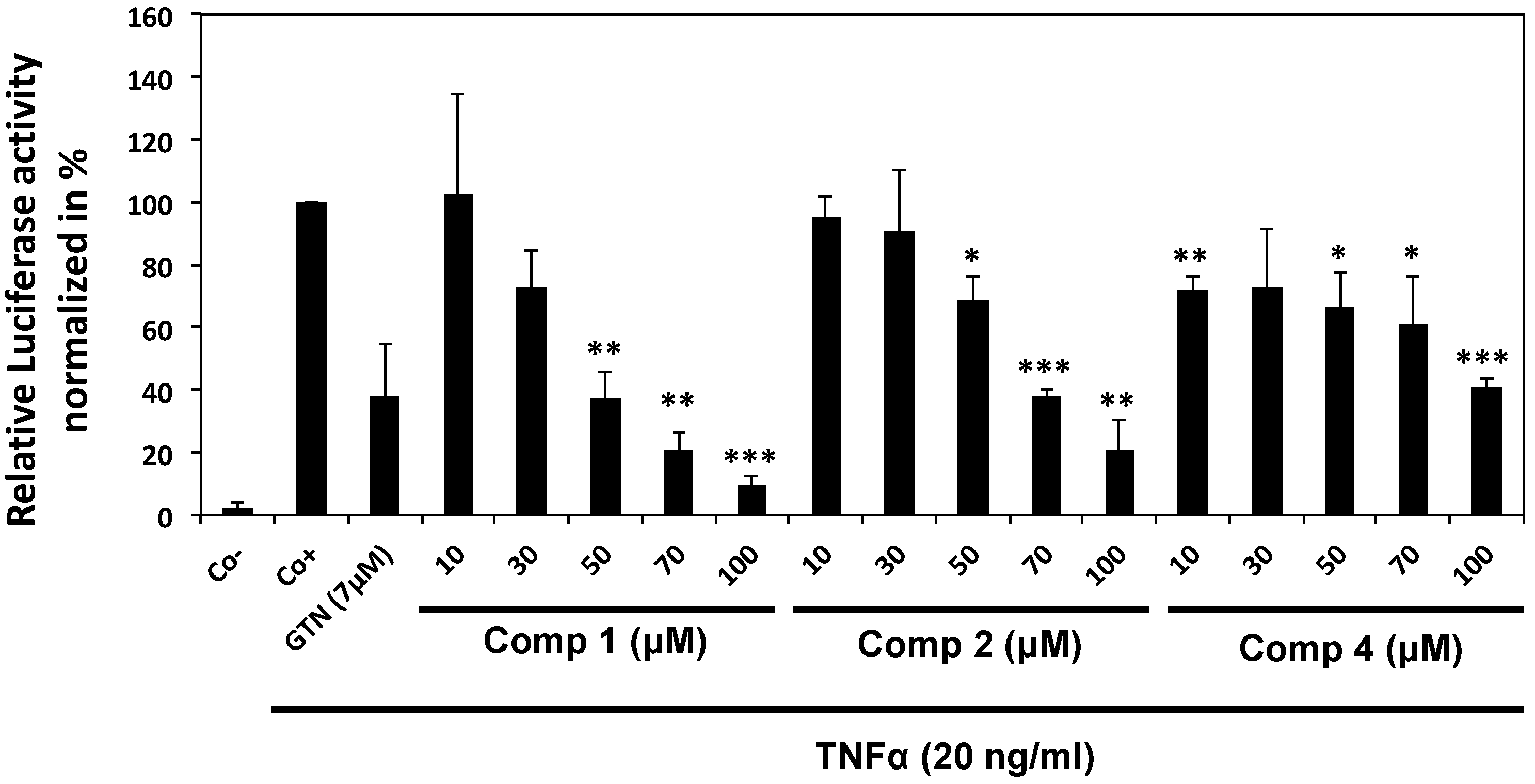
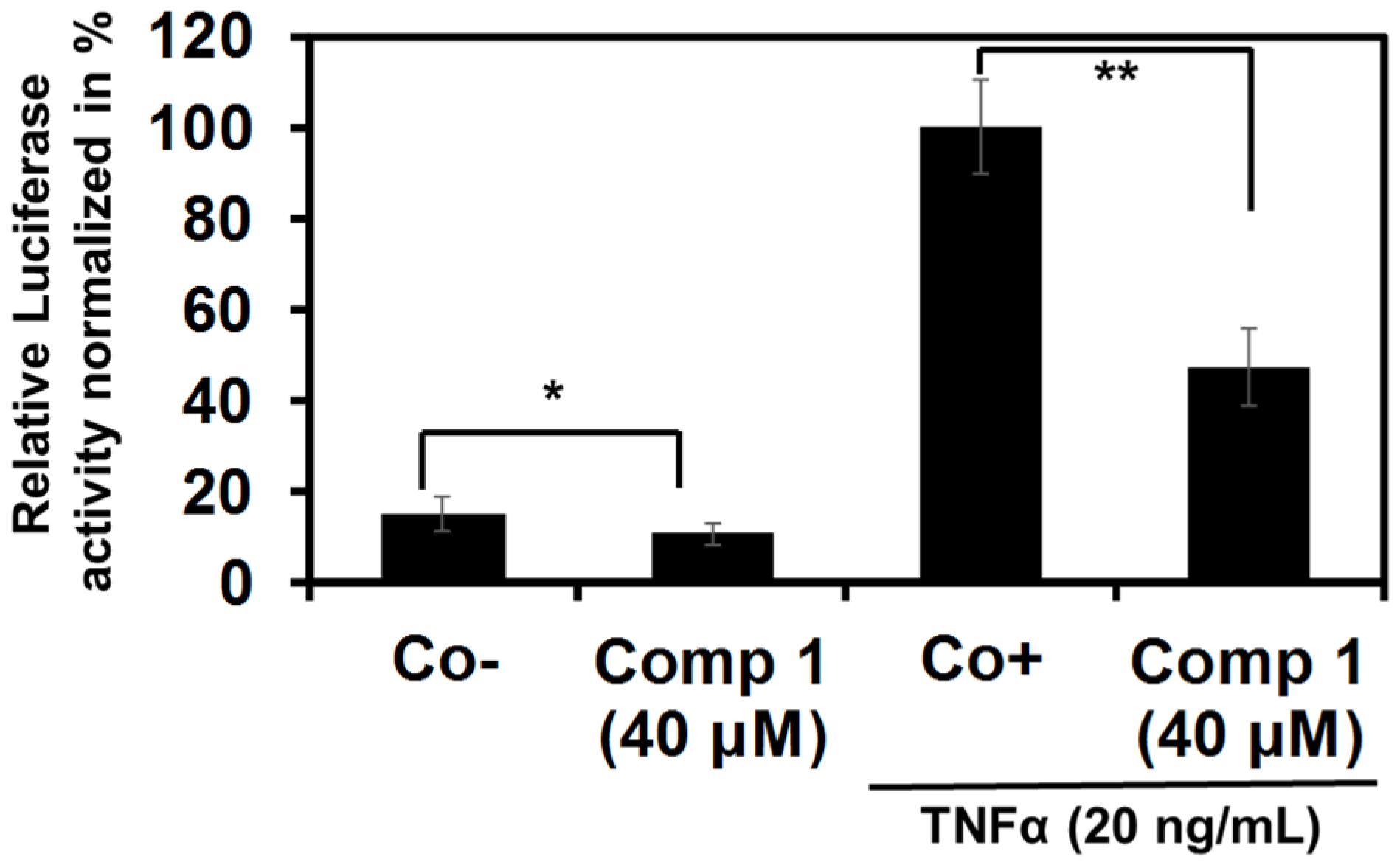
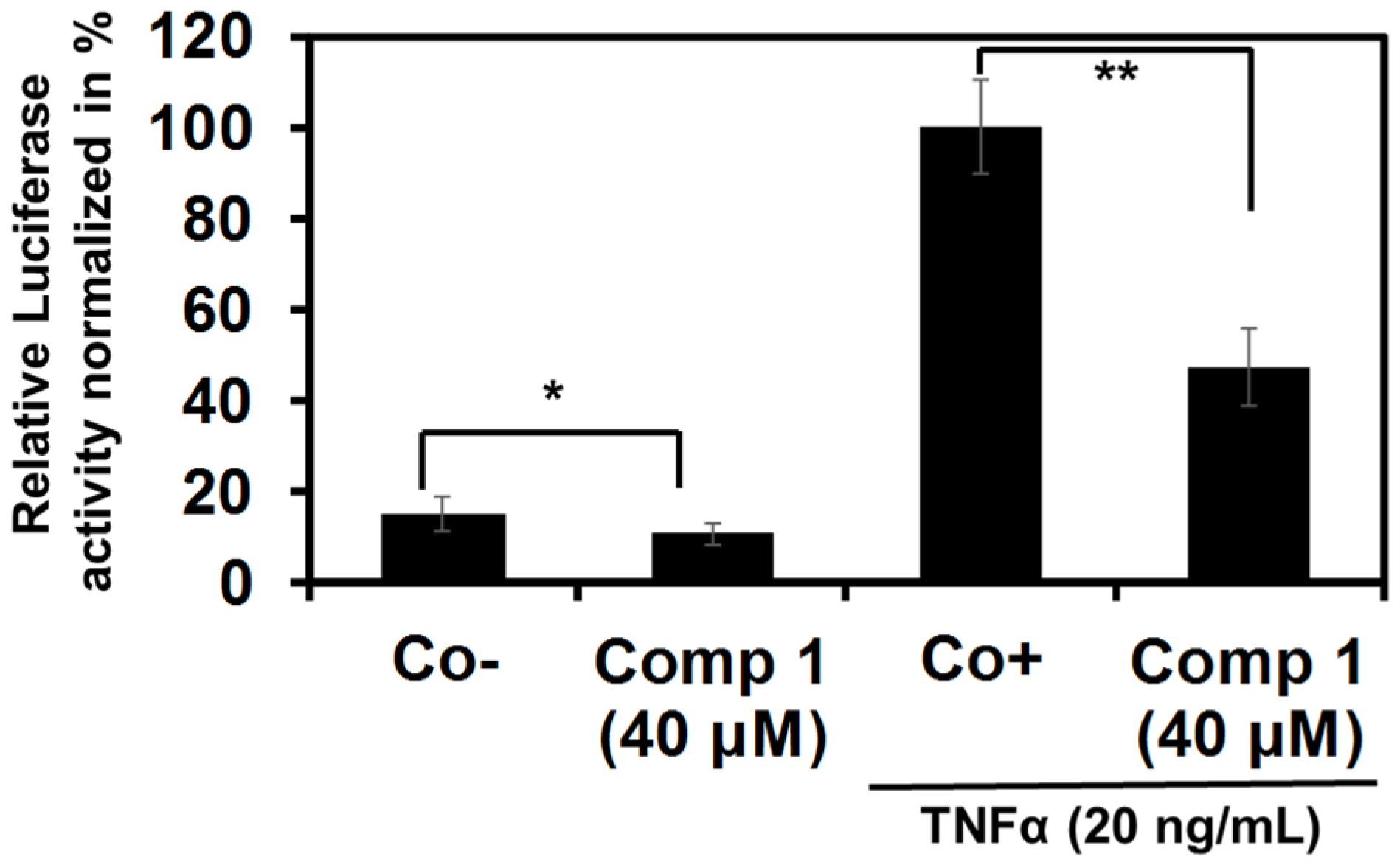
2.2.1. Epipolythiodiketopiperazines Dose-Dependently Inhibit TNF Alpha-Induced NF-κB Activation

2.2.2. 6-Acetylmonodethiogliotoxin Down-Regulates the Expression of NF-κB Target Genes

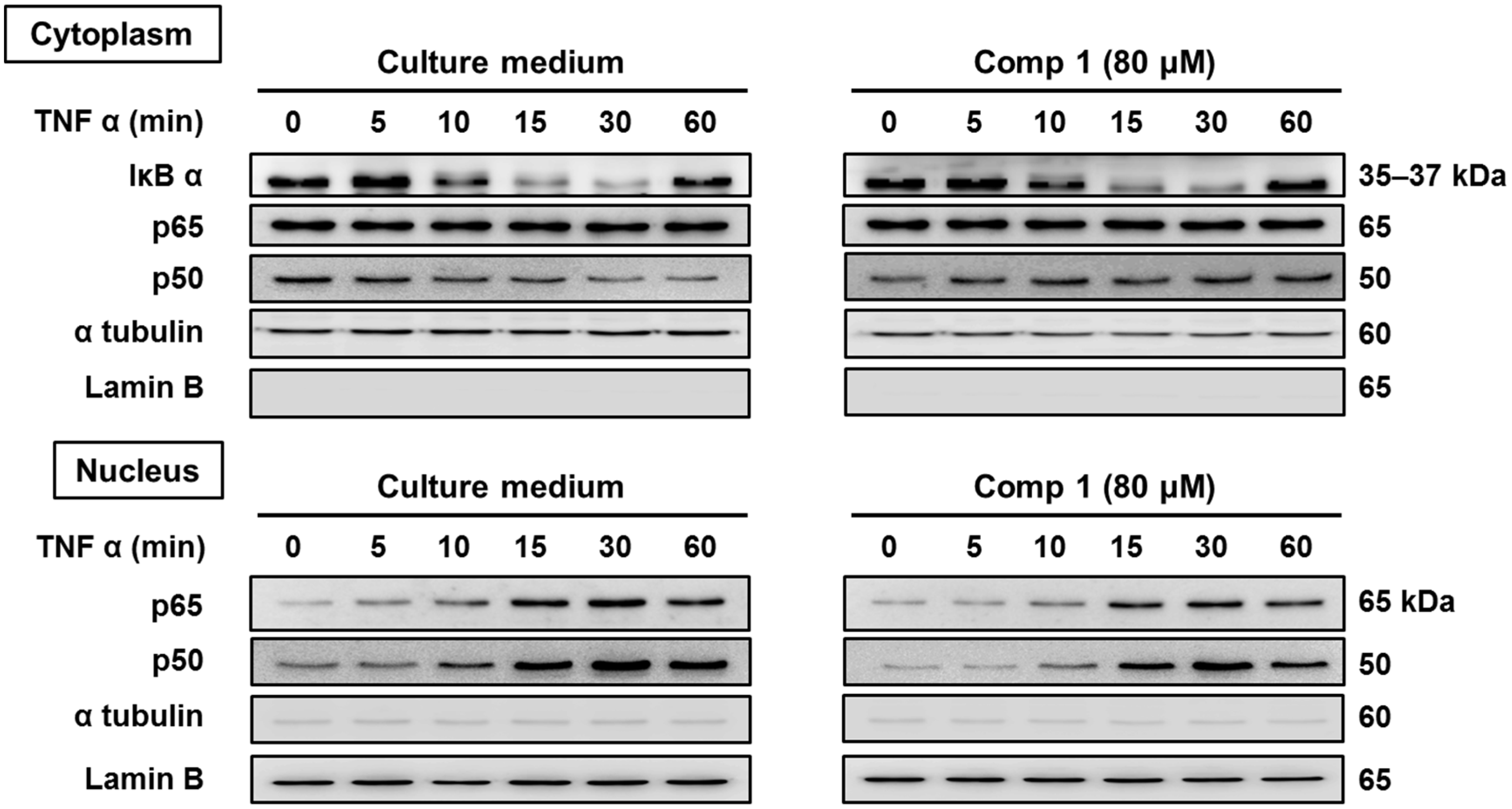
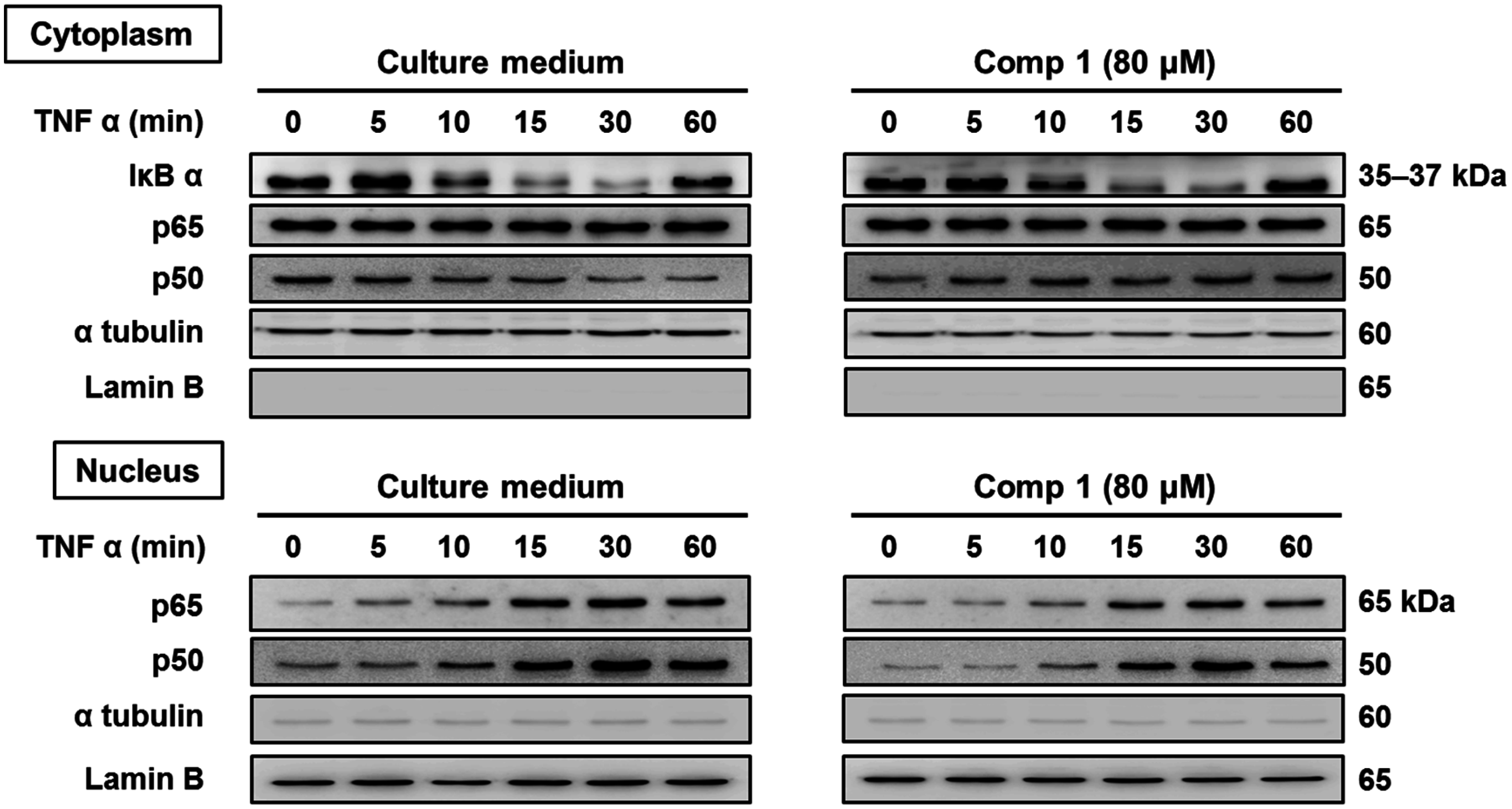
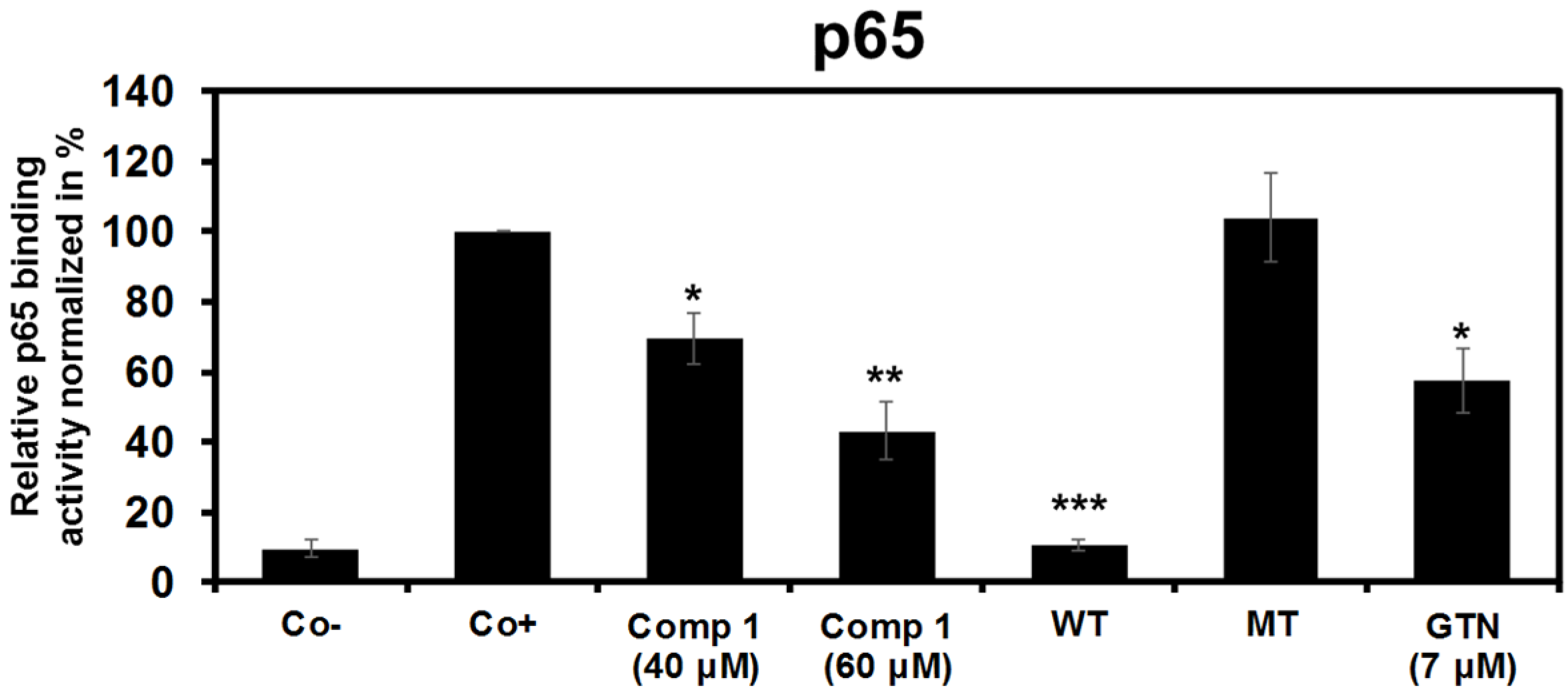
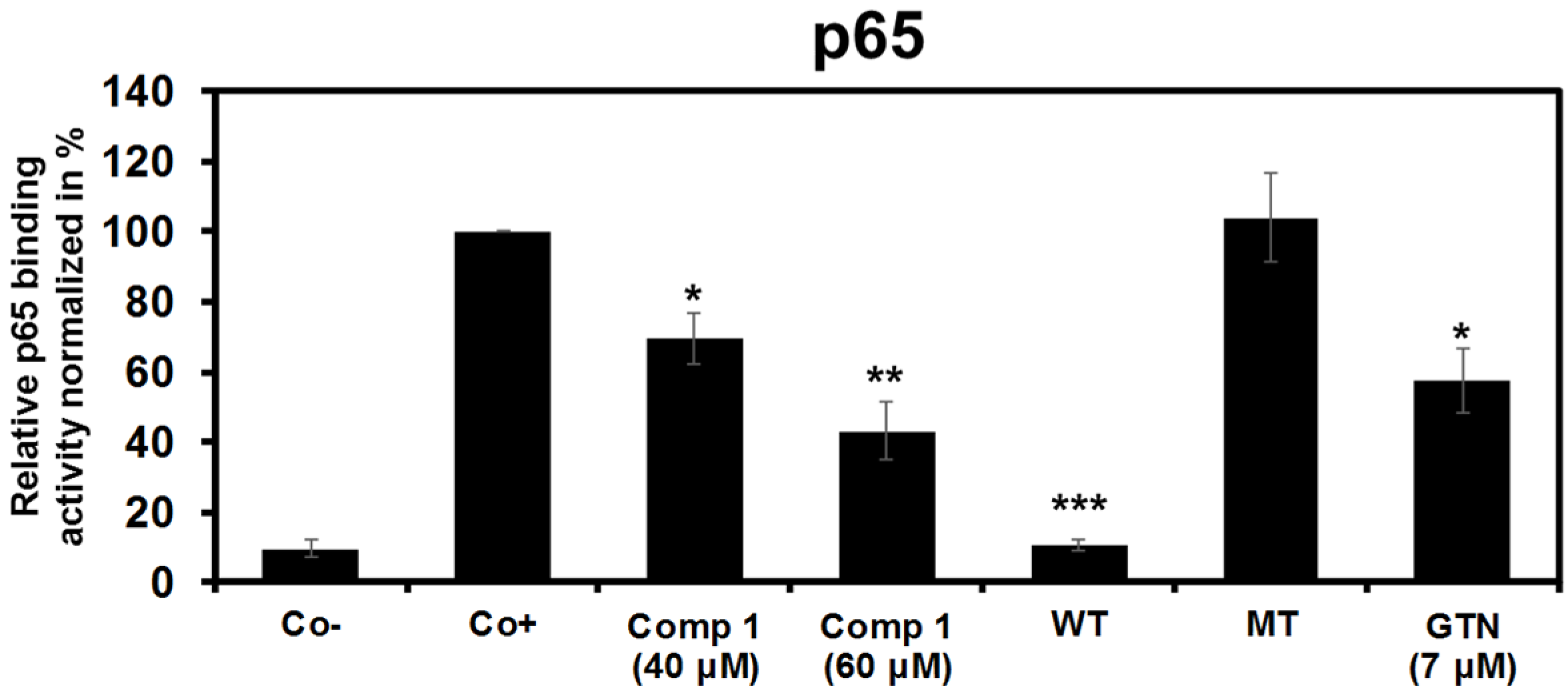
2.2.3. 6-Acetylmonodethiogliotoxin Mediated Downstream Inhibition of NF-κB Signaling by Preventing Binding of p65 to DNA



2.3. Discussion
3. Experimental Section
3.1. General Procedures for Natural Products Chemistry
3.2. Cultivation of Fungus, Extraction and Isolation
3.3. Bioassays
3.3.1. Cell Culture
3.3.2. Transient Transfection and Luciferase Reporter Gene Assay
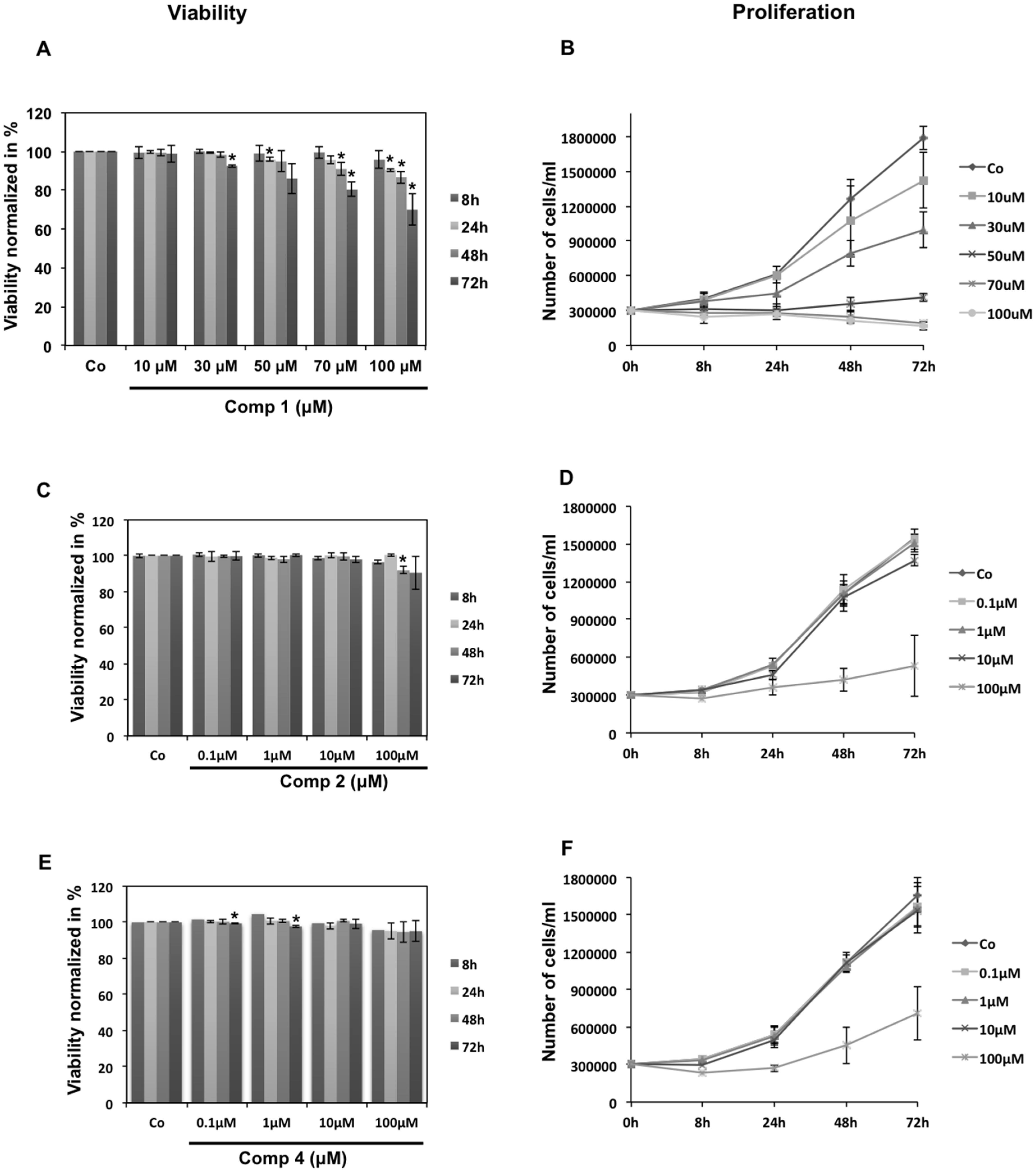
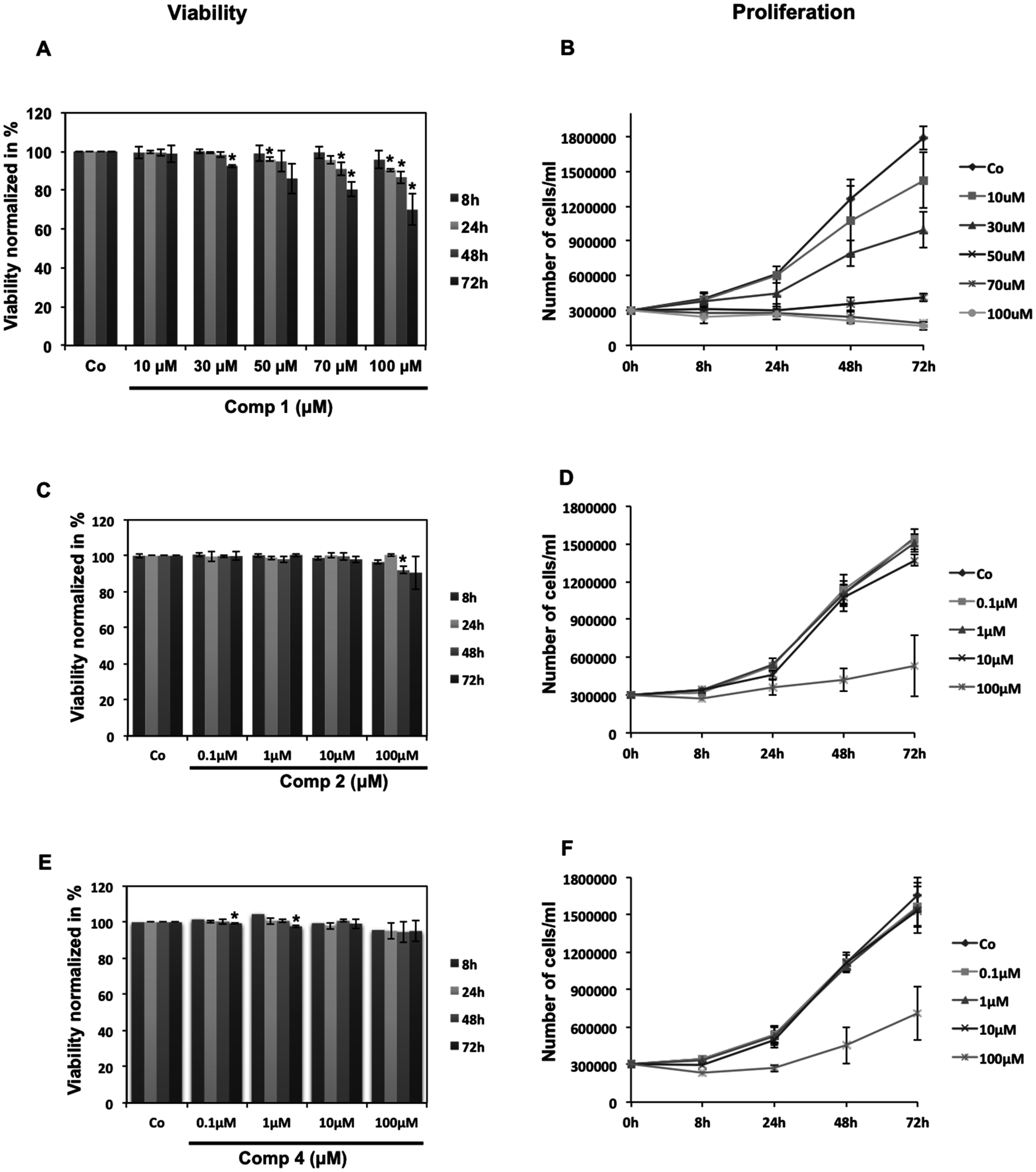
3.3.3. Cell Viability Assessment
3.3.4. Extraction of Cellular Proteins
3.3.5. Western Blot Analysis
3.3.6. TransAM Assay
3.3.7. Statistical Analysis
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- König, G.M.; Kehraus, S.; Seibert, S.F.; Abdel-Lateff, A.; Müller, D. Natural products from marine organisms and their associated microbes. ChemBioChem 2006, 7, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Epigenetic modulators from “The Big Blue”: A treasure to fight against cancer. Cancer Lett. 2014, 351, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.H.; Debbab, A.; Clements, C.; Edrada-Ebel, R.; Orlikova, B.; Diederich, M.; Wray, V.; Lin, W.; Proksch, P. NF kappa B inhibitors and antitrypanosomal metabolites from endophytic fungus Penicillium sp. isolated from Limonium tubiflorum. Bioorganic Med. Chem. 2011, 19, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Panwalkar, A.; Verstovsek, S.; Giles, F. Nuclear factor-kappaB modulation as a therapeutic approach in hematologic malignancies. Cancer 2004, 100, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.E.; Huang, D.B.; Chen, Y.Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Tasdemir, D.; Golais, F.; Dicato, M.; Diederich, M. The aromatic ketone 4'-hydroxychalcone inhibits TNFalpha-induced NF-kappaB activation via proteasome inhibition. Biochem. Pharmacol. 2011, 82, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, T.; Nakatsuka, S.I.; Kishi, Y. Total synthesis of gliotoxin, dehydrogliotoxin and hyalodendrin. Tetrahedron 1981, 37, 2045–2078. [Google Scholar] [CrossRef]
- Johnson, J.R.; Bruce, W.F.; Dutcher, J.D. Gliotoxin, The Antibiotic Principle of Gliocladium fimbriatum. I. Production, Physical and Biological Properties1. J. Am. Chem. Soc. 1943, 65, 2005–2009. [Google Scholar] [CrossRef]
- Kaouadji, M.; Steiman, R.; Seigle-Murandi, F.; Krivobok, S.; Sage, L. Gliotoxin: Uncommon 1H couplings and revised 1H-and 13C-NMR assignments. J. Nat. Prod. 1990, 53, 717–719. [Google Scholar] [CrossRef]
- Scharf, D.H.; Chankhamjon, P.; Scherlach, K.; Heinekamp, T.; Willing, K.; Brakhage, A.A.; Hertweck, C. Epidithiodiketopiperazine Biosynthesis: A Four-Enzyme Cascade Converts Glutathione Conjugates into Transannular Disulfide Bridges. Angew. Chem. Intl. Ed. 2013, 52, 11092–11095. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.M.; Waring, P.; Howlett, B.J. The epipolythiodioxopiperazine (ETP) class of fungal toxins: Distribution, mode of action, functions and biosynthesis. Microbiology 2005, 151, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.; Fridrichsons, J.; Mathieson, A.M. The structure and absolute configuration of gliotoxin and the absolute configuration of sporidesmin. Tetrahedron Lett. 1966, 7, 3131–3138. [Google Scholar] [CrossRef]
- Watts, K.R.; Ratnam, J.; Ang, K.H.; Tenney, K.; Compton, J.E.; McKerrow, J.; Crews, P. Assessing the trypanocidal potential of natural and semi-synthetic diketopiperazines from two deep water marine-derived fungi. Bioorganic Med. Chem. 2010, 18, 2566–2574. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Yoshida, K.; Uchida, I.; Nishikawa, M.; Kohsaka, M.; Aoki, H. Studies of platelet activating factor (PAF) antagonists from microbial products. I. Bisdethiobis (methylthio) gliotoxin and its derivatives. Chem. Pharm. Bull. 1986, 34, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Hosoe, T.; Fukushima, K.; Itabashi, T.; Nozawa, K.; Takizawa, K.; Kawai, K.I. The absolute structures of dihydroepiheveadride, as characteristic antifungal agent against filamentous fungi, and its related compounds from unidentified fungus IFM 52672. Heterocycles Sendai Inst. Heterocycl. Chem. 2004, 63, 2581–2590. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Gehlot, P. Inflammation and cancer: How friendly is the relationship for cancer patients? Curr. Opin. Pharmacol. 2009, 9, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Hajjouli, S.; Chateauvieux, S.; Teiten, M.H.; Orlikova, B.; Schumacher, M.; Dicato, M.; Choo, C.Y.; Diederich, M. Eurycomanone and eurycomanol from Eurycoma longifolia Jack as regulators of signaling pathways involved in proliferation, cell death and inflammation. Molecules 2014, 19, 14649–14666. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Legrand, N.; Panning, J.; Dicato, M.; Diederich, M. Anti-inflammatory and anticancer drugs from nature. Cancer Treatment Res. 2014, 159, 123–143. [Google Scholar]
- Orhan, I.E.; Kartal, M.; Gulpinar, A.R.; Yetkin, G.; Orlikova, B.; Diederich, M.; Tasdemir, D. Inhibitory effect of St. Johns Wort oil macerates on TNF-α-induced NF-κB activation and their fatty acid composition. J. Ethnopharmacol. 2014, 155, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Schumacher, M.; Juncker, T.; Yan, C.C.; Inayat-Hussain, S.H.; Hajjouli, S.; Cerella, C.; Dicato, M.; Diederich, M. Styryl-lactone goniothalamin inhibits TNF-α-induced NF-κB activation. Food Chem. Toxicol. 2013, 59, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, W.; Aly, A.H.; Wray, V.; Mandi, A.; Teiten, M.H.; Gaascht, F.; Orlikova, B.; Kassack, M.U.; Lin, W.; Diederich, M.; et al. Embellicines A and B: Absolute configuration and NF-κB transcriptional inhibitory activity. J. Med. Chem. 2013, 56, 2991–2999. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Taylor, A. Sporidesmins. Part XI. The reaction of triphenylphosphine with epipolythiodioxopiperazines. J. Chem. Soc. C Org. 1971, 3, 1189–1192. [Google Scholar] [CrossRef]
- Pedras, M.S.C.; Abrams, S.R.; Séguin-Swartz, G. Isolation of the first naturally occurring epimonothiodioxopiperazine, a fungal toxin produced by Phoma lingam. Tetrahedron Lett. 1988, 29, 3471–3474. [Google Scholar] [CrossRef]
- Herath, H.M.; Jacob, M.; Wilson, A.D.; Abbas, H.K.; Nanayakkara, N.P. New secondary metabolites from bioactive extracts of the fungus Armillaria tabescens. Nat. Prod. Res. 2013, 27, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Crane, R.I.; Hedden, P.; MacMillan, J.; Turner, W.B. Fungal products. IV. The structure of heveadride, a new nonadrede from Helminthosporium heveae. J. Chem. Soc. Perkin Trans. 1 1973, 2, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Duvoix, A.; Delhalle, S.; Blasius, R.; Schnekenburger, M.; Morceau, F.; Fougere, M.; Henry, E.; Galteau, M.M.; Dicato, M.; Diederich, M. Effect of chemopreventive agents on glutathione S-transferase P1-1 gene expression mechanisms via activating protein 1 and nuclear factor kappa B inhibition. Biochem. Pharmacol. 2004, 68, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harms, H.; Orlikova, B.; Ji, S.; Nesaei-Mosaferan, D.; König, G.M.; Diederich, M. Epipolythiodiketopiperazines from the Marine Derived Fungus Dichotomomyces cejpii with NF-κB Inhibitory Potential. Mar. Drugs 2015, 13, 4949-4966. https://doi.org/10.3390/md13084949
Harms H, Orlikova B, Ji S, Nesaei-Mosaferan D, König GM, Diederich M. Epipolythiodiketopiperazines from the Marine Derived Fungus Dichotomomyces cejpii with NF-κB Inhibitory Potential. Marine Drugs. 2015; 13(8):4949-4966. https://doi.org/10.3390/md13084949
Chicago/Turabian StyleHarms, Henrik, Barbora Orlikova, Seungwon Ji, Damun Nesaei-Mosaferan, Gabriele M. König, and Marc Diederich. 2015. "Epipolythiodiketopiperazines from the Marine Derived Fungus Dichotomomyces cejpii with NF-κB Inhibitory Potential" Marine Drugs 13, no. 8: 4949-4966. https://doi.org/10.3390/md13084949
APA StyleHarms, H., Orlikova, B., Ji, S., Nesaei-Mosaferan, D., König, G. M., & Diederich, M. (2015). Epipolythiodiketopiperazines from the Marine Derived Fungus Dichotomomyces cejpii with NF-κB Inhibitory Potential. Marine Drugs, 13(8), 4949-4966. https://doi.org/10.3390/md13084949