
Kunitz-Type Peptide HCRG21 from the Sea Anemone Heteractis crispa Is a Full Antagonist of the TRPV1 Receptor

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
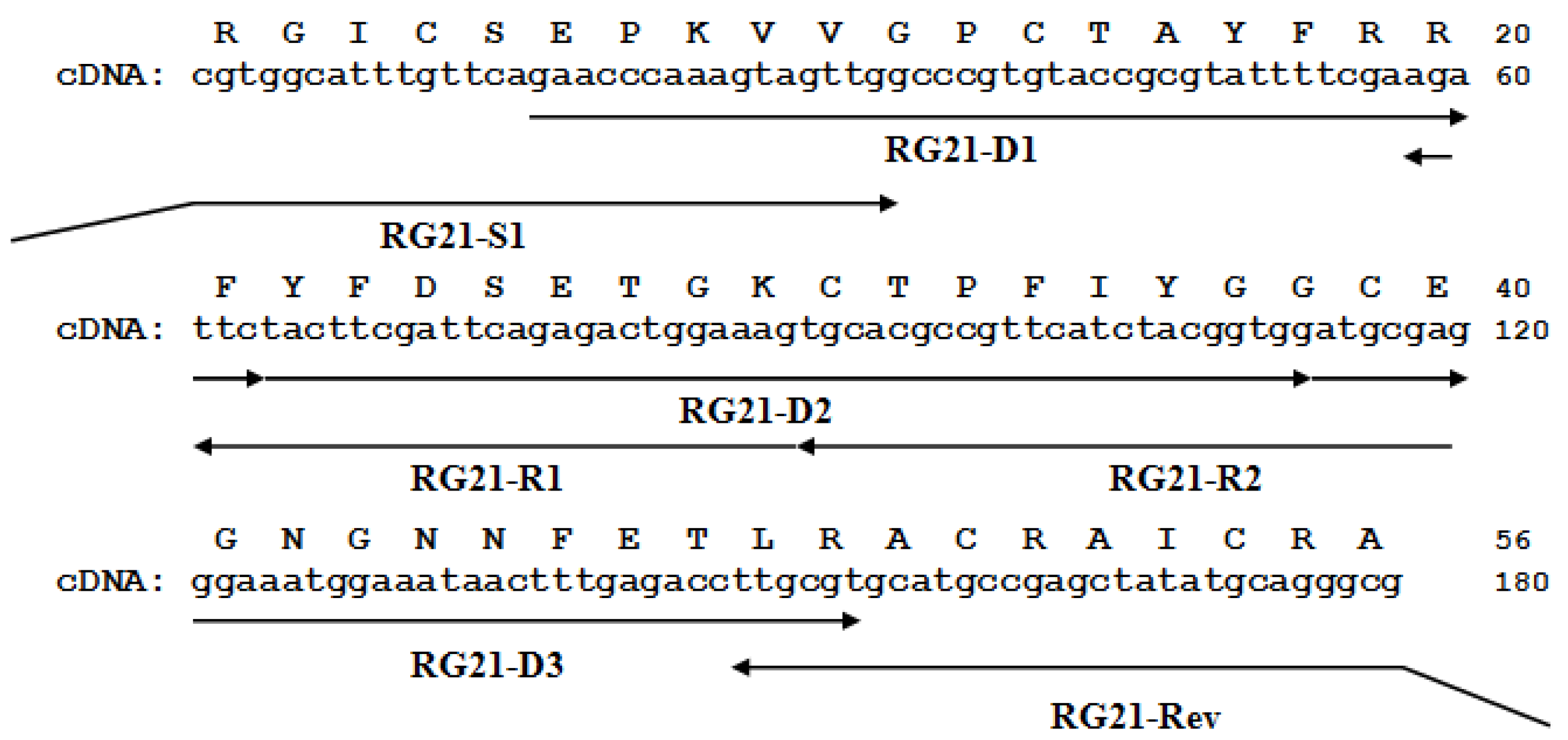
2.1. cDNA hcrg21 Gene and Recombinant Peptide Obtaining
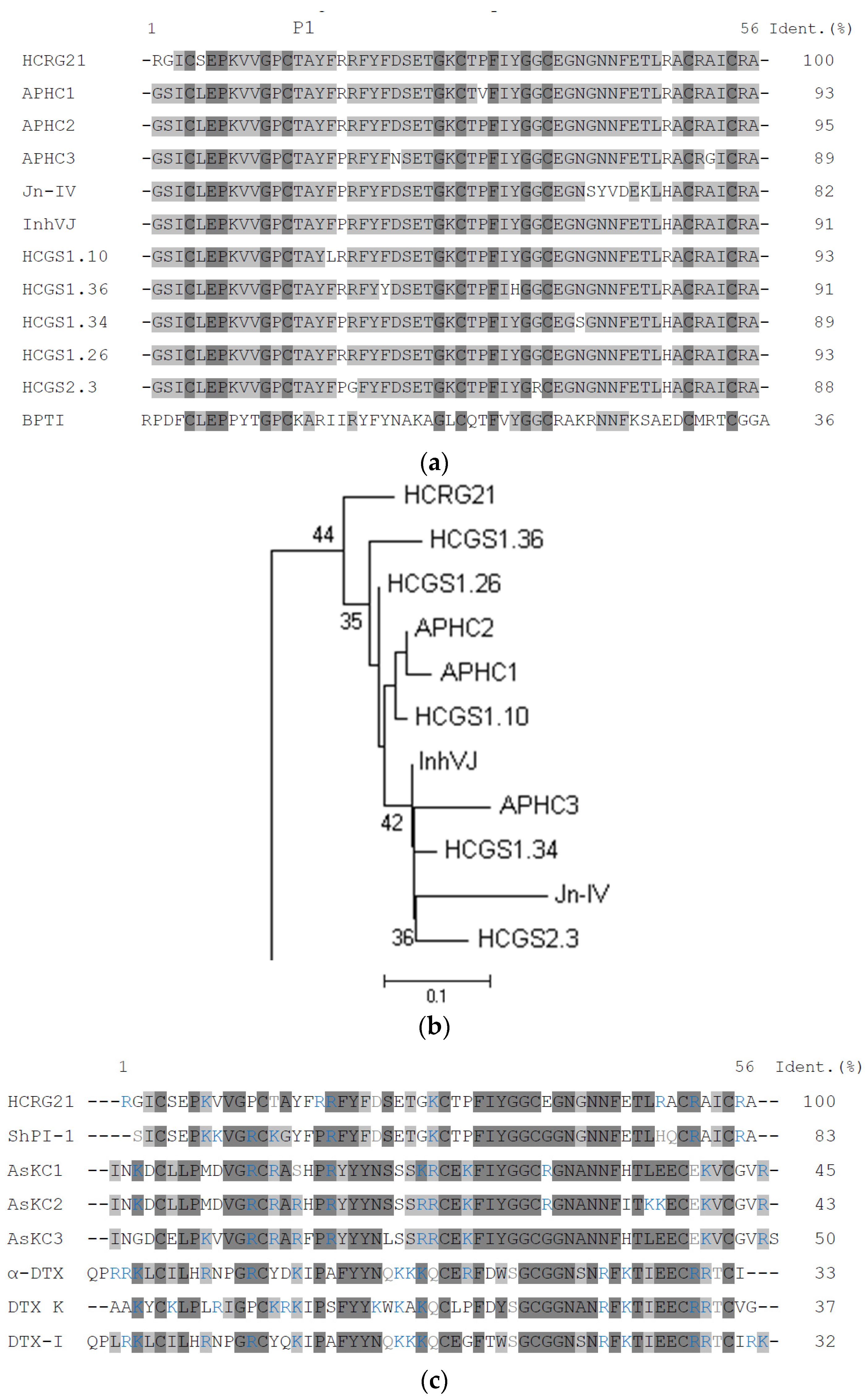
2.2. Comparison of Kunitz-Type Inhibitors Protease Inhibiting Activity
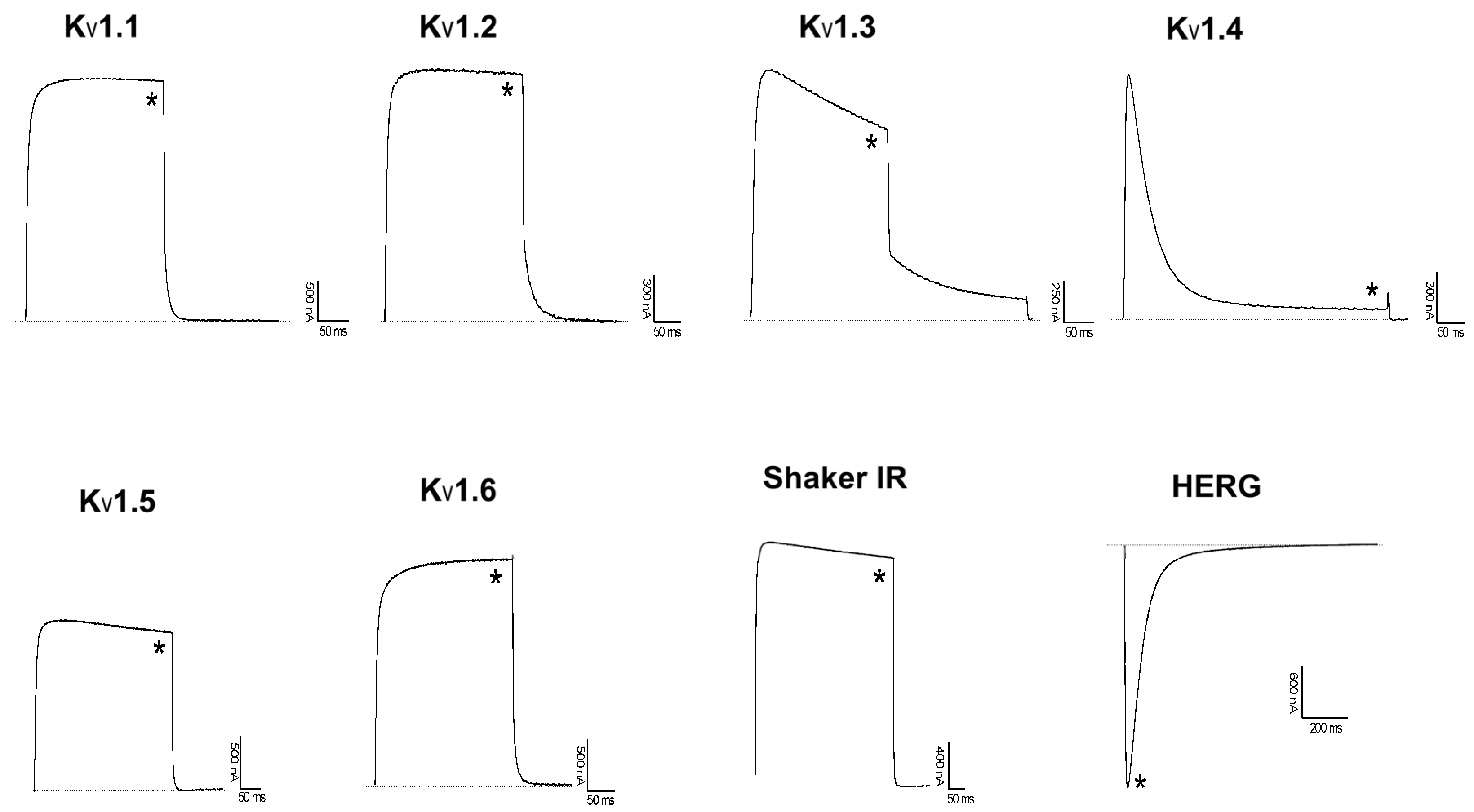
2.3. Activity to Kv and TRPV1 Channels: Electrophysiological Assay and Comparative Structure Analysis
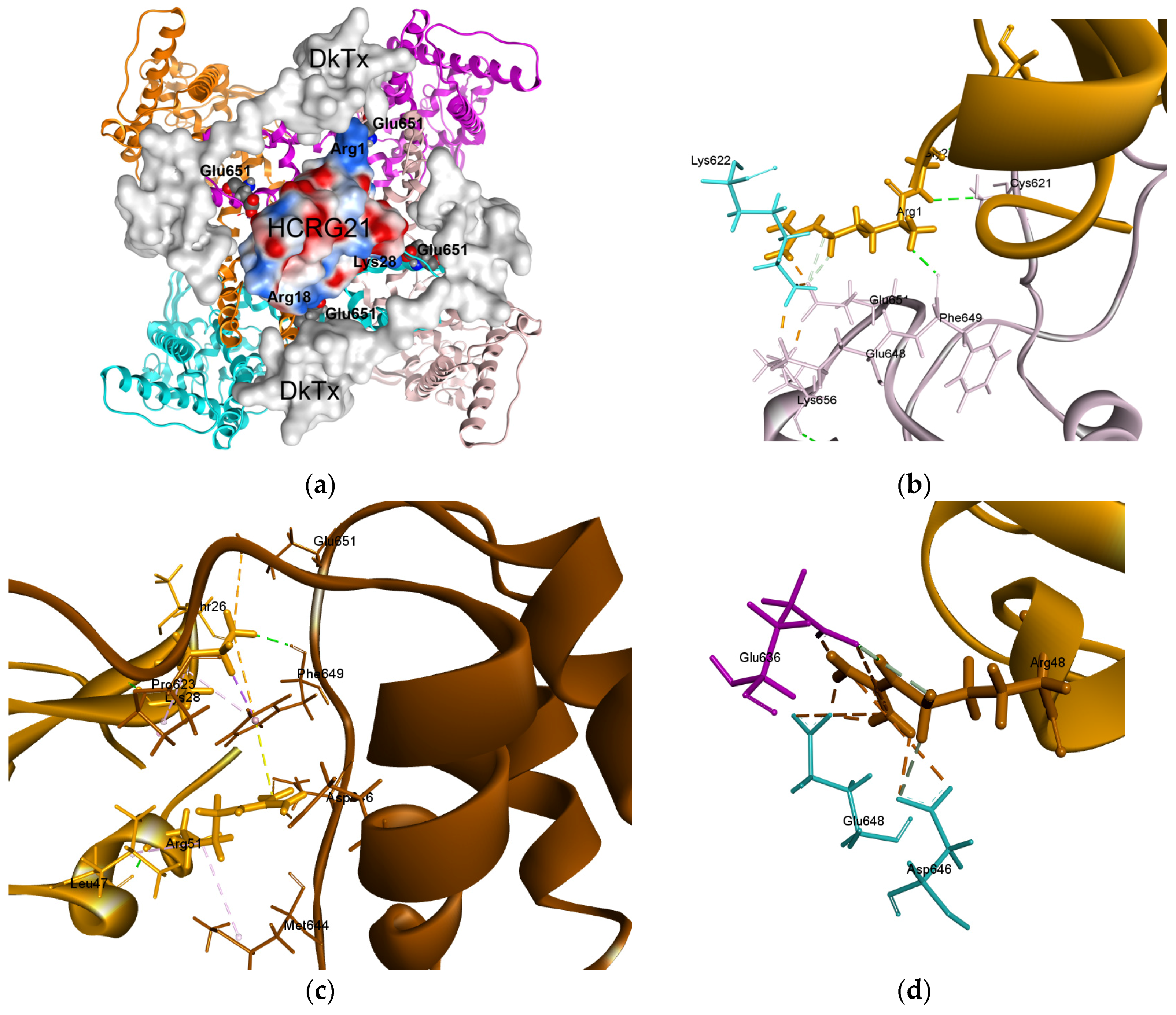
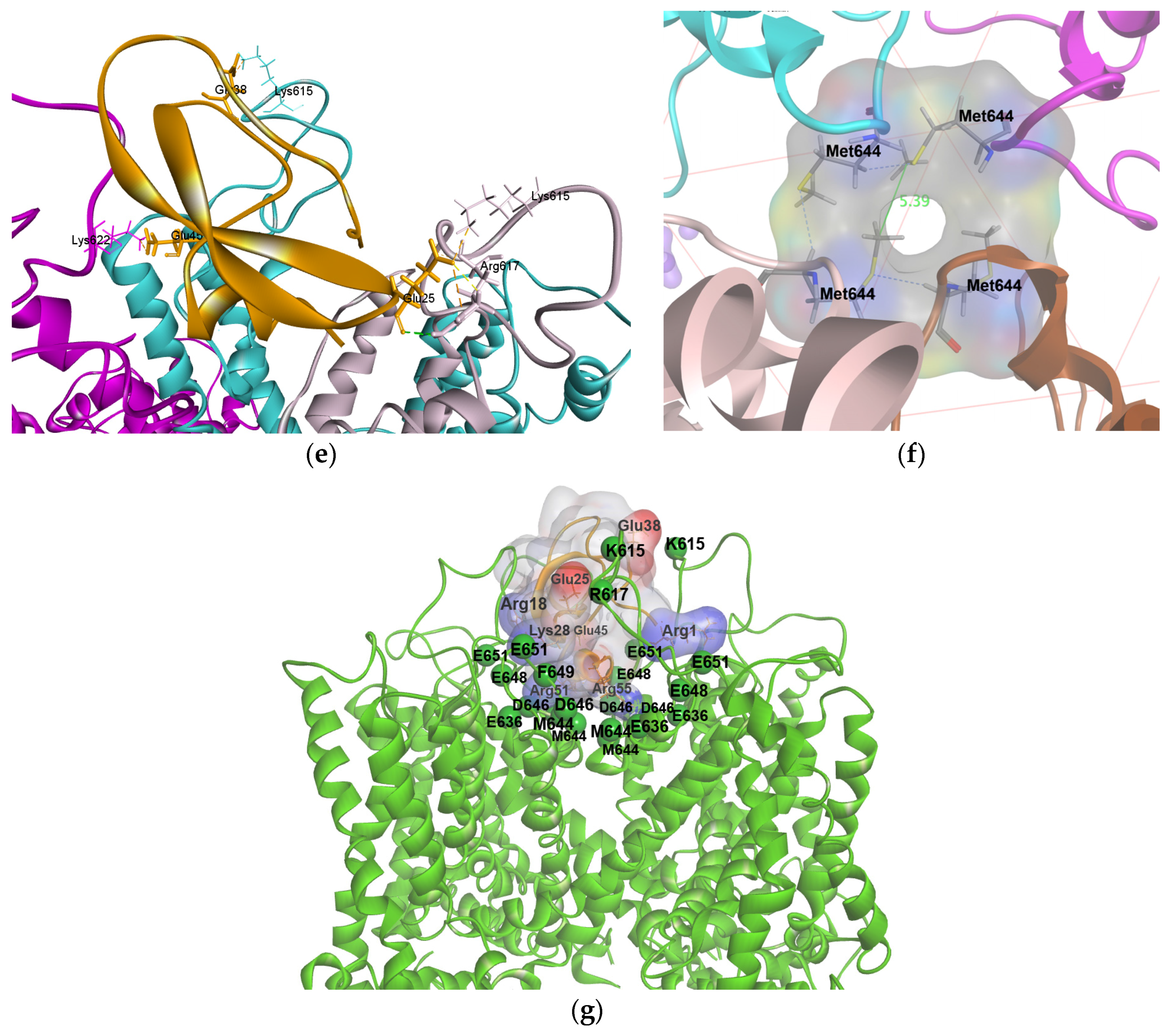
2.4. Molecular Modeling
3. Materials and Methods
3.1. Gene Sequence Determination
3.2. Synthesis of Gene Encoding HCRG21
3.3. Production and Physical and Chemical Characterization of Recombinant HCRG21
3.4. Phylogenetic Analysis
3.5. Inhibitory Activity
3.6. Expression of Voltage-Gated Ion Channels in Xenopus laevis Oocytes
3.7. Electrophysiological Recordings
3.8. Homology Modeling and Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.; King, G.F.; Nevalainen, T.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef] [PubMed]
- Jouiaei, M.; Yanagihara, A.A.; Madio, B.; Nevalainen, T.J.; Alewood, P.F.; Fry, B.G. Ancient Venom Systems: A Review on Cnidaria Toxins. Toxins 2015, 7, 2251–2271. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Kalia, J.; Milescu, M.; Salvatierra, J.; Wagner, J.; Klint, J.K.; King, G.F.; Olivera, B.M.; Bosmans, F. From foe to friend: Using animal toxins to investigate ion channel function. J. Mol. Biol. 2015, 427, 158–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazao, B.; Vasconcelos, V.; Antunes, A. Sea anemone (cnidaria, anthozoa, actiniaria) toxins: An overview. Mar. Drugs 2012, 10, 1812–1851. [Google Scholar] [CrossRef] [PubMed]
- Mourão, C.B.F.; Schwartz, E.F. Protease inhibitors from marine venomous animals and their counterparts in terrestrial venomous animals. Mar. Drugs 2013, 11, 2069–2112. [Google Scholar] [CrossRef] [PubMed]
- Schweitz, H.; Bruhn, T.; Guillemare, E.; Moinier, D.; Lancelin, J.M.; Beress, L.; Lazdunski, M. Kalicludines and kaliseptine. Two different classes of sea anemone toxins for voltage sensitive K+ channels. J. Biol. Chem. 1995, 270, 25121–25126. [Google Scholar] [CrossRef] [PubMed]
- Honma, T.; Kawahata, S.; Ishida, M.; Nagai, H.; Nagashima, Y.; Shiomi, K. Novel peptide toxins from the sea anemone Stichodactyla haddoni. Peptides 2008, 29, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Billen, B.; Derua, R.; Waelkens, E.; Debaveye, S.; Beress, L.; Tytgat, J. A bifunctional sea anemone peptide with Kunitz type protease and potassium channel inhibiting properties. Biochem. Pharmacol. 2011, 82, 81–90. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, R.; Peigneur, S.; Pons, T.; Alvarez, C.; González, L.; Chávez, M.A.; Tytgat, J. The Kunitz-type protein ShPI-1 inhibits serine proteases and Voltage-Gated potassium channels. Toxins 2016, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Isaeva, M.P.; Chausova, V.E.; Zelepuga, E.A.; Guzev, K.V.; Tabakmakher, V.M.; Monastyrnaya, M.M.; Kozlovskaya, E.P. A new multigene superfamily of Kunitz-type protease inhibitors from sea anemone Heteractis crispa. Peptides 2012, 34, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Steele, R.E.; David, C.N.; Technau, U. A genomic view of 500 million years of cnidarian evolution. Trends Genet. 2011, 27, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Wunderer, G.; Machleidt, W.; Fritz, H. The broad-specificity proteinase inhibitor 5 II from the sea anemone Anemonia sulcata. Methods Enzymol. 1981, 88, 816–820. [Google Scholar]
- Zykova, T.A.; Vinokurov, L.M.; Markova, L.F.; Kozlovskaya, E.P.; Elyakov, G.B. Amino-acid sequence of trypsin inhibitor IV from Radianthus macrodactylus. Bioorg. Khim. 1985, 11, 293–301. [Google Scholar]
- Sokotun, I.N.; Il’ina, A.P.; Monastyrnaya, M.M.; Leychenko, E.V.; Es’kov, A.A.; Anastuk, S.D.; Kozlovskaya, E.P. Proteinase inhibitors from the tropical sea anemone Radianthus macrodactylus: Isolation and characteristic. Biochemistry (Mosc.) 2007, 72, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Sokotun, I.N.; Leychenko, E.V.; Vakorina, T.I.; Es’kov, A.A.; Il’ina, A.P.; Monastyrnaia, M.M.; Kozlovskaia, E.P. A serine protease inhibitor from the anemone Radianthus macrodactylus: Isolation and physicochemical characteristics. Russ. J. Bioorg. Chem. 2007, 33, 415–422. [Google Scholar] [CrossRef]
- Gladkikh, I.; Monastyrnaya, M.; Leychenko, E.; Zelepuga, E.; Chausova, V.; Isaeva, M.; Anastyuk, S.; Andreev, Y.; Peigneur, S.; Tytgat, J. Atypical reactive center Kunitz-type inhibitor from the sea anemone Heteractis crispa. Mar. Drugs 2012, 10, 1545–1565. [Google Scholar] [CrossRef] [PubMed]
- Gladkikh, I.; Monastyrnaya, M.; Zelepuga, E.; Sintsova, O.; Tabakmakher, V.; Gnedenko, O.; Ivanov, A.; Hua, K.-F.; Kozlovskaya, E. New Kunitz-type HCRG polypeptides from the sea anemone Heteractis crispa. Mar. Drugs 2015, 13, 6038–6063. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, S.; Ishida, M.; Shimakura, K.; Nagashima, Y.; Shiomi, K. Isolation and amino acid sequences of two Kunitz-type protease inhibitors from the sea anemone Anthopleura aff. xanthogrammica. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997, 118, 381–386. [Google Scholar] [CrossRef]
- Minagawa, S.; Ishida, M.; Shimakura, K.; Nagashima, Y.; Shiomi, K. Amino acid sequence and biological activities of another Kunitz-type protease inhibitor from the sea anemone Anthopleura aff. xanthogrammica. Fish. Sci. 1998, 64, 155–159. [Google Scholar]
- Minagawa, S.; Sugiyama, M.; Ishida, M.; Nagashima, Y.; Shiomi, K. Kunitz-type protease inhibitors from acrorhagi of three species of sea anemones. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 150, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Delfín, J.; Martínez, I.; Antuch, W.; Morera, V.; González, Y.; Rodríguez, R.; Márquez, M.; Saroyán, A.; Larionova, N.; Díaz, J.; et al. Purification, characterization and immobilization of proteinase inhibitors from Stichodactyla helianthus. Toxicon 1996, 34, 1367–1376. [Google Scholar] [CrossRef]
- Díaz, J.; Morera, V.; Delfín, J.; Huerta, V.; Lima, G.; Rodriguex de la Vega, M.; Garcia, B.; Padrón, G.; Assfalg-Machleidt, I.; Machleidt, W.; et al. Purification and partial characterization of a novel proteinase inhibitor from the sea anemone Stichodactyla helianthus. Toxicon 1998, 36, 1275–1276. [Google Scholar]
- Antuch, W.; Berndt, D.K.; Chávez, A.M.; Delfín, J.; Wüthrich, K. The NMR solution structure of a Kunitz-type proteinase inhibitor from the sea anemone Stichodactyla helianthus. Eur. J. Biochem. 1993, 212, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Tabakmakher, V.M.; Sintsova, O.V.; Krivoshapko, O.N.; Zelepuga, E.A.; Monastyrnaya, M.M.; Kozlovskaya, E.P. Analgesic effect of novel Kunitz-type polypeptides of the sea anemone Heteractis crispa. Dokl. Biochem. Biophys. (Russ.) 2015, 461, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Sintsova, O.V.; Monastyrnaya, M.M.; Pislyagin, E.A.; Menchinskaya, E.S.; Leychenko, E.V.; Aminin, D.L.; Kozlovskaya, E.P. Anti-Inflammatory Activity of a Polypeptide from the Heteractis crispa Sea Anemone. Russ. J. Bioorg. Chem. 2015, 41, 590–596. [Google Scholar] [CrossRef]
- Sintsova, O.V.; Pislyagin, E.A.; Gladkikh, I.N.; Monastyrnaya, M.M.; Menchinskaya, E.S.; Leychenko, E.V.; Aminin, D.L.; Kozlovskaya, E.P. Kunitz-type peptides of the sea anemone Heteractis crispa—Potential anti-inflammatory compounds. Russ. J. Bioorg. Chem. 2015, 13, 6038–6063. [Google Scholar]
- Andreev, Y.A.; Kozlov, S.A.; Koshelev, S.G.; Ivanova, E.A.; Monastyrnaya, M.M.; Kozlovskaya, E.P.; Grishin, E.V. Analgesic compound from sea anemone Heteractis crispa is the first polypeptide inhibitor of vanilloid receptor 1 (TRPV1). J. Biol. Chem. 2008, 283, 23914–23921. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.A.; Andreev, Y.A.; Murashev, A.N.; Skobtsov, D.I.; D’yachenko, I.A.; Grishin, E.V. New polypeptide components from the Heteractis crispa sea anemone with analgesic activity. Russ. J. Bioorg. Chem. 2009, 35, 711–719. [Google Scholar] [CrossRef]
- Andreev, Y.A.; Kozlov, S.A.; Korolkova, Y.V.; Dyachenko, I.A.; Bondarenko, D.A.; Skobtsov, D.I.; Murashev, A.N.; Kotova, P.D.; Rogachevskaja, O.A.; Kabanova, N.V.; et al. Polypeptide modulators of TRPV1 produce analgesia without hyperthermia. Mar. Drugs 2013, 11, 5100–5115. [Google Scholar] [CrossRef] [PubMed]
- Dyachenko, I.A.; Andreev, Y.A.; Logashina, Yu.A.; Murashev, A.N.; Grishin, E.V. Biological Activity of a Polypeptide Modulator of TRPV1 Receptor. Dokl. Biol. Sci. (Russ.) 2015, 465, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Zelepuga, E.A.; Tabakmakher, V.M.; Chausova, V.E.; Monastyrnaya, M.M.; Isaeva, M.P.; Kozlovskaya, E.P. Interaction of sea anemone Heteractis crispa Kunitz type polypeptides with pain vanilloid receptor TRPV1: In silico investigation. Russ. J. Bioorg. Chem. 2012, 38, 159–170. [Google Scholar] [CrossRef]
- Salat, K.; Moniczewski, A.; Librowski, T. Transient receptor potential channels—Emerging novel drug targets for the treatment of pain. Curr. Med. Chem. 2013, 20, 1409–1436. [Google Scholar] [CrossRef] [PubMed]
- Julius, D. TRP channels and pain. Annu. Rev. Cell Dev. Biol. 2013, 29, 355–384. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A. Transient receptor potential channels in pain and inflammation: Therapeutic opportunities. Pain Pract. 2010, 10, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Jung, S.J.; Oh, S.B. Role of TRP channels in pain sensation. Adv. Exp. Med. Biol. 2011, 704, 615–636. [Google Scholar] [PubMed]
- Venkatachalam, K.; Montell, C. TRP Channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Molecular mechanism of TRP channels. Compr. Physiol. 2013, 3, 221–242. [Google Scholar] [PubMed]
- Cui, M.; Honore, P.; Zhong, C.; Gauvin, D.; Mikusa, J.; Hernandez, G.; Chandran, P.; Gomtsyan, A.; Brown, B.; Bayburt, E.K.; et al. TRPV1 receptors in the CNS play a key role in broad-spectrum analgesia of TRPV1 antagonists. J. Neurosci. 2006, 26, 9385–9393. [Google Scholar] [CrossRef] [PubMed]
- Khairatkar-Joshi, N.; Szallasi, A. TRPV1 antagonists: The challenges for therapeutic targeting. Trends Mol. Med. 2009, 15, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.Y.; Gavva, N.R. Therapeutic potential of vanilloid receptor TRPV1 agonists and antagonists as analgesics: Recent advances and setbacks. Brain Res. Rev. 2009, 60, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, M.J.; Szallasi, A. Peripheral TRPV1 receptors as targets for drug development: New molecules and mechanisms. Curr. Pharm. Des. 2008, 14, 32–41. [Google Scholar] [PubMed]
- Chizh, B.A.; O’Donnell, M.B.; Napolitano, A.; Wang, J.; Brooke, A.C.; Aylott, M.C.; Bullman, J.N.; Gray, E.J.; Lai, R.Y.; Williams, P.M.; et al. The effects of the TRPV1 antagonist SB-705498 on TRPV1 receptor-mediated activity and inflammatory hyperalgesia in humans. Pain 2007, 132, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Weber, U.J.; Gotfredsen, J.W.; Jayatissa, M.; Buus, C.; Kristensen, N.B.; Vestergaard, M.; Teschendorf, P.; Schneider, A.; Hansen, P.; et al. Drug-induced mild therapeutic hypothermia obtained by administration of a transient receptor potential vanilloid type 1 agonist. BMC Cardiovasc. Disord. 2010, 10, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, V.; Rohacs, T. TRPV1: A Target for Rational Drug Design. Pharmaceuticals 2016, 9, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, M.J.; Chizh, B.A. Clinical development of TRPV1 antagonists: Targeting a pivotal point in the pain pathway. Drug Discov. Today 2009, 14, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Priel, A.; Zhou, S.; King, D.; Siemens, J.; Julius, D. A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 2010, 141, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Siemens, J.; Zhou, S.; Piskorowski, R.; Nikai, T.; Lumpkin, E.A.; Basbaum, A.I.; King, D.; Julius, D. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006, 444, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Hakim, M.A.; Jiang, W.; Luo, L.; Li, B.; Yang, S.; Song, Y.; Lai, R. Scorpion toxin, BmP01, induces pain by targeting TRPV1 channel. Toxins 2015, 7, 3671–3687. [Google Scholar] [CrossRef] [PubMed]
- Monastyrnaya, M.; Chausova, V.; Isaeva, M.; Gladkikh, I.; Zelepuga, E.; Leychenko, E.; Kozlovskaya, E.P. New Kunitz-type peptide HCRG subfamily from the sea anemone. Heteractis crispa. In preparation.
- Harvey, A.L. Twenty years of dendrotoxins. Toxicon 2001, 39, 15–26. [Google Scholar] [CrossRef]
- Robertson, B.; Owen, D.; Stow, J.; Butler, C.; Newland, C. Novel effects of dendrotoxin homologues on subtypes of mammalian Kv1 potassium channels expressed in Xenopus oocytes. FEBS Lett. 1996, 383, 26–30. [Google Scholar] [CrossRef]
- Owen, D.G.; Hall, A.; Stephens, G.; Stow, J.; Robertson, B. The relative potencies of dendrotoxins as blockers of the cloned voltage-gated K+ channel, mKv1.1 (MK-1), when stably expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 1997, 120, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Kassell, B. Bovine Trypsin-Kallikrein Inhibitor (Kunitz Inhibitor, Basic Pancreatic Trypsin Inhibitor, Polyvalent Inhibitor from Bovine Organs). In Methods in Enzymology; Lorand, L., Ed.; Academic Press: San Francisco, CA, USA, 1970; Volume 19, pp. 844–852. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Huger, R. Structural basis of the endoproteinase-protein inhibitor interaction. Biochim. Biophys. Acta 2000, 1477, 241–252. [Google Scholar] [CrossRef]
- Helland, R.; Otlewski, J.; Sundheim, O.; Dadlez, M.; Smalås, A.O. The crystal structures of the complexes between bovine beta-trypsin and ten P1 variants of BPTI. J. Mol. Biol. 1999, 287, 923–942. [Google Scholar] [CrossRef] [PubMed]
- Czapinska, H.; Helland, R.; Smalås, A.O.; Otlewski, J. Crystal structures of five bovine chymotrypsin complexes with P1 BPTI variants. J. Mol. Biol. 2004, 344, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Fernández Ballester, G.; Ferrer Montiel, A. Molecular modeling of the full-length human TRPV1 channel in closed and desensitized states. J. Membr. Biol. 2008, 223, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Lazar, J.; Braun, D.C.; Tóth, A.; Wang, Y.; Pearce, L.V.; Pavlyukovets, V.A.; Blumberg, P.M.; Garfield, S.H.; Wincovitch, S.; Choi, H.K.; et al. Kinetics of penetration influence the apparent potency of vanilloids on TRPV1. Mol. Pharmacol. 2006, 69, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, C.; Morenilla-Palao, C.; Planells-Cases, R.; Merino, J.M.; Ferrer-Montiel, A. Identification of an aspartic residue in the P-loop of the vanilloid receptor that modulates pore properties. J. Biol. Chem. 2000, 275, 32552–32558. [Google Scholar] [CrossRef] [PubMed]
- Chugunov, A.O.; Volynsky, P.E.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. Temperature-sensitive gating of TRPV1 channel as probed by atomistic simulations of its trans- and juxtamembrane domains. Sci. Rep. 2016, 6, 33112. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Gosu, V.; Basith, S.; Hong, S.; Choi, S. Polymodal Transient Receptor Potential Vanilloid Type 1 Nocisensor: Structure, Modulators, and Therapeutic Applications. Adv. Protein Chem. Struct. Biol. 2016, 104, 81–125. [Google Scholar] [PubMed]
- Munns, C.H.; Chung, M.K.; Sanchez, Y.E.; Amzel, L.M.; Caterina, M.J. Role of the outer pore domain in transient receptor potential vanilloid 1 dynamic permeability to large cations. J. Biol. Chem. 2015, 290, 5707–5724. [Google Scholar] [CrossRef] [PubMed]
- Caires, R.; Luis, E.; Taberner, F.J.; Fernandez-Ballester, G.; Ferrer-Montiel, A.; Balazs, E.A.; Gomis, A.; Belmonte, C.; de la Peña, E. Hyaluronan modulates TRPV1 channel opening, reducing peripheral nociceptor activity and pain. Nat. Commun. 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Yamaguchi, Y.; Kawano, C.; Nose, T.; Nirthanan, S.; Gopalakrishnakone, P.; Tytgat, J.; Sato, K. Active sites of Spinoxin, a potassium channel scorpion toxin, elucidated by systematic alanine scanning. Biochemistry 2016, 55, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Il’ina, A.; Lipkin, A.; Barsova, E.; Issaeva, M.; Leychenko, E.; Guzev, K.; Monastyrnaya, M.; Lukyanov, S.; Kozlovskaya, E. Amino acid sequence of RTX-A’s isoform actinoporins from the sea anemone, Radianthus macrodactylus. Toxicon 2006, 47, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russel, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; pp. 33–36. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Andreev, Y.A.; Kozlov, S.A.; Vassilevski, A.A.; Grishin, E.V. Cyanogen bromide cleavage of proteins in salt and buffer solutions. Anal. Biochem. 2010, 407, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Zuckerkand, E.; Pauling, L. Evolutionary Divergence and Convergence in Proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: San Francisco, CA, USA, 1965; pp. 97–166. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Dixon, M.; Webb, E.C. Enzymes; Mir Publishing: Moscow, Russia, 1982; Volume 2, pp. 397–661. [Google Scholar]
- Liman, E.R.; Tytgat, J.; Hess, P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 1992, 9, 861–871. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [PubMed]
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016.
- Laskowski, R.A.; Rullman, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Siemens, J.; Hanack, C. Modulation of TRP Ion Channels by Venomous Toxins. In Book Mammalian Transient Receptor Potential (TRP) Cation Channels; Nilius, B., Flockerzi, V., Eds.; Springer International Publishing: Cham, Switzerland, 2014; Volume 2, pp. 1119–1142. [Google Scholar]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins Struct. Funct. Bioinform. 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins Struct. Funct. Bioinform. 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. Amber 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2016; Dassault Systèmes: San Diego, CA, USA, 2016.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sea Anemone | Peptide | Mr, Da | P1 Residue | Ki, M | |
|---|---|---|---|---|---|
| Trypsin | α-Chymotrypsin | ||||
| Bos taurus | BPTI [55] | K | 6.0 × 10−14 | 1.8 × 10−13 | |
| A. sulcata | AsKC1 [7] | 6685 * | R | <3 × 10−10 | n.d. |
| AsKC2 [7] | 6772 * | <3 × 10−10 | n.d. | ||
| AsKC3 [7] | 6732 * | <3 × 10−10 | n.d. | ||
| SA5 II [13] | 6937 ** | 3.0 × 10−10 | n.d. | ||
| A. elegantissima | APEKTx1 [9] | 7469 * | 1.2 × 10−7 * | n.d. | |
| S. helianthus | SHPI-1 [23] | 6110 * | K | 1.1 × 10−10 | 2.3 × 10−9 |
| SHPI-2 [23] | 6195 ** | 3.8 × 10−10 | n.d. | ||
| H. crispa | HCRG1 [18] | 6196 * | 2.8 × 10−8 | n.d. | |
| HCRG2 [18] | 6148.7 * | 5.0 × 10−8 | n.d. | ||
| H. crispa | rHCRG21 | 6228 * | T | 2.0 × 10−7 | 7.0 × 10−7 |
| Jn-IV [14] | 6165 * | 9.6 × 10−9 | n.d. | ||
| InhVJ [17] | 6106 * | 7.38 × 10−8 | 9.93 × 10−7 | ||
| APHC1 [28] | 6187 * | 1.0 × 10−6 | 5.0 × 10−6 | ||
| APHC2 [29] | 6185 * | 9.0 × 10−7 | 4.5 × 10−6 | ||
| APHC3 [29] | 6111 * | 5.0 × 10−7 | 7.0 × 10−6 | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monastyrnaya, M.; Peigneur, S.; Zelepuga, E.; Sintsova, O.; Gladkikh, I.; Leychenko, E.; Isaeva, M.; Tytgat, J.; Kozlovskaya, E. Kunitz-Type Peptide HCRG21 from the Sea Anemone Heteractis crispa Is a Full Antagonist of the TRPV1 Receptor. Mar. Drugs 2016, 14, 229. https://doi.org/10.3390/md14120229
Monastyrnaya M, Peigneur S, Zelepuga E, Sintsova O, Gladkikh I, Leychenko E, Isaeva M, Tytgat J, Kozlovskaya E. Kunitz-Type Peptide HCRG21 from the Sea Anemone Heteractis crispa Is a Full Antagonist of the TRPV1 Receptor. Marine Drugs. 2016; 14(12):229. https://doi.org/10.3390/md14120229
Chicago/Turabian StyleMonastyrnaya, Margarita, Steve Peigneur, Elena Zelepuga, Oksana Sintsova, Irina Gladkikh, Elena Leychenko, Marina Isaeva, Jan Tytgat, and Emma Kozlovskaya. 2016. "Kunitz-Type Peptide HCRG21 from the Sea Anemone Heteractis crispa Is a Full Antagonist of the TRPV1 Receptor" Marine Drugs 14, no. 12: 229. https://doi.org/10.3390/md14120229
APA StyleMonastyrnaya, M., Peigneur, S., Zelepuga, E., Sintsova, O., Gladkikh, I., Leychenko, E., Isaeva, M., Tytgat, J., & Kozlovskaya, E. (2016). Kunitz-Type Peptide HCRG21 from the Sea Anemone Heteractis crispa Is a Full Antagonist of the TRPV1 Receptor. Marine Drugs, 14(12), 229. https://doi.org/10.3390/md14120229