
Prospecting Biotechnologically-Relevant Monooxygenases from Cold Sediment Metagenomes: An In Silico Approach

 and
and 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Identification of Metagenomic Sequences
2.2. BVMOs
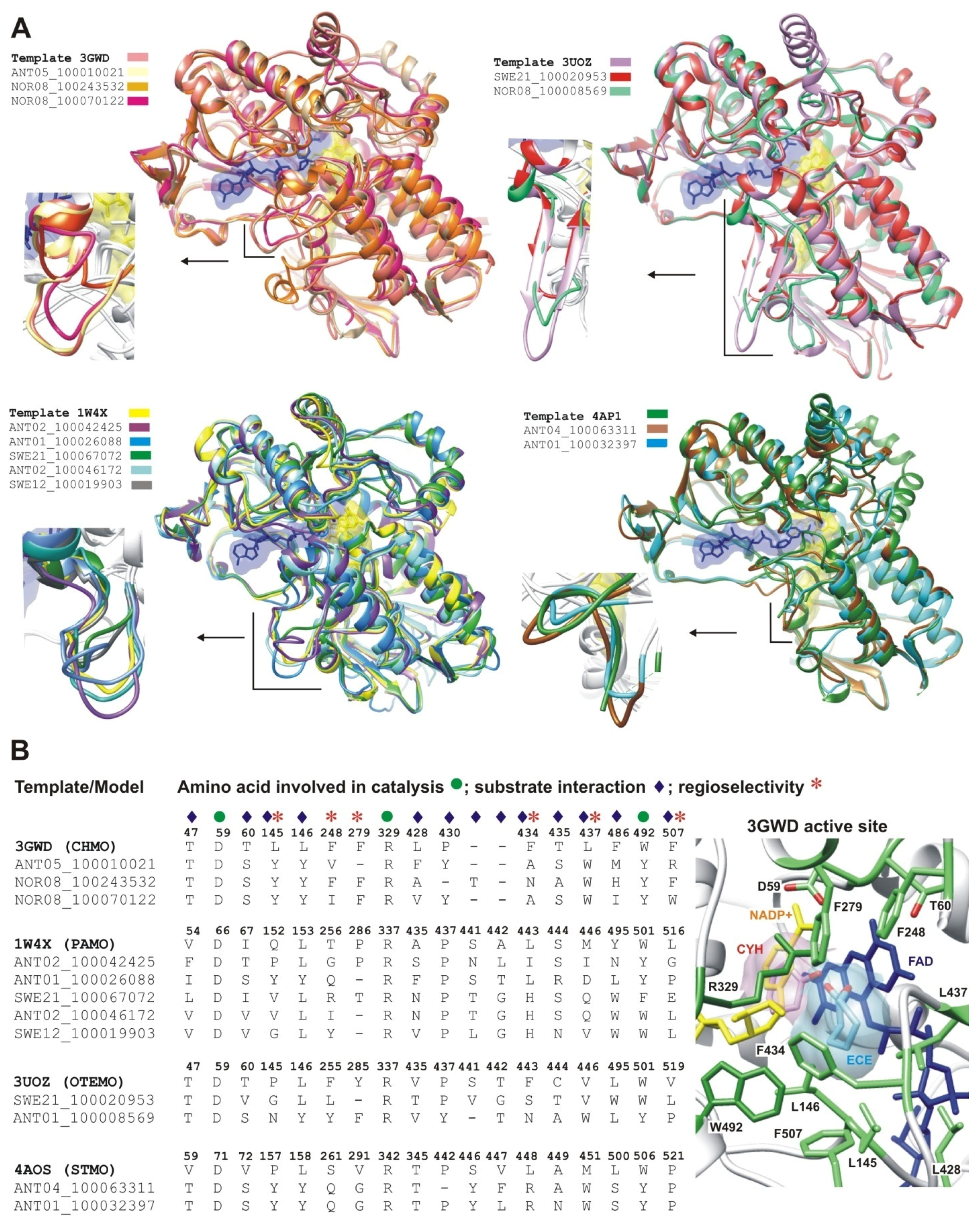
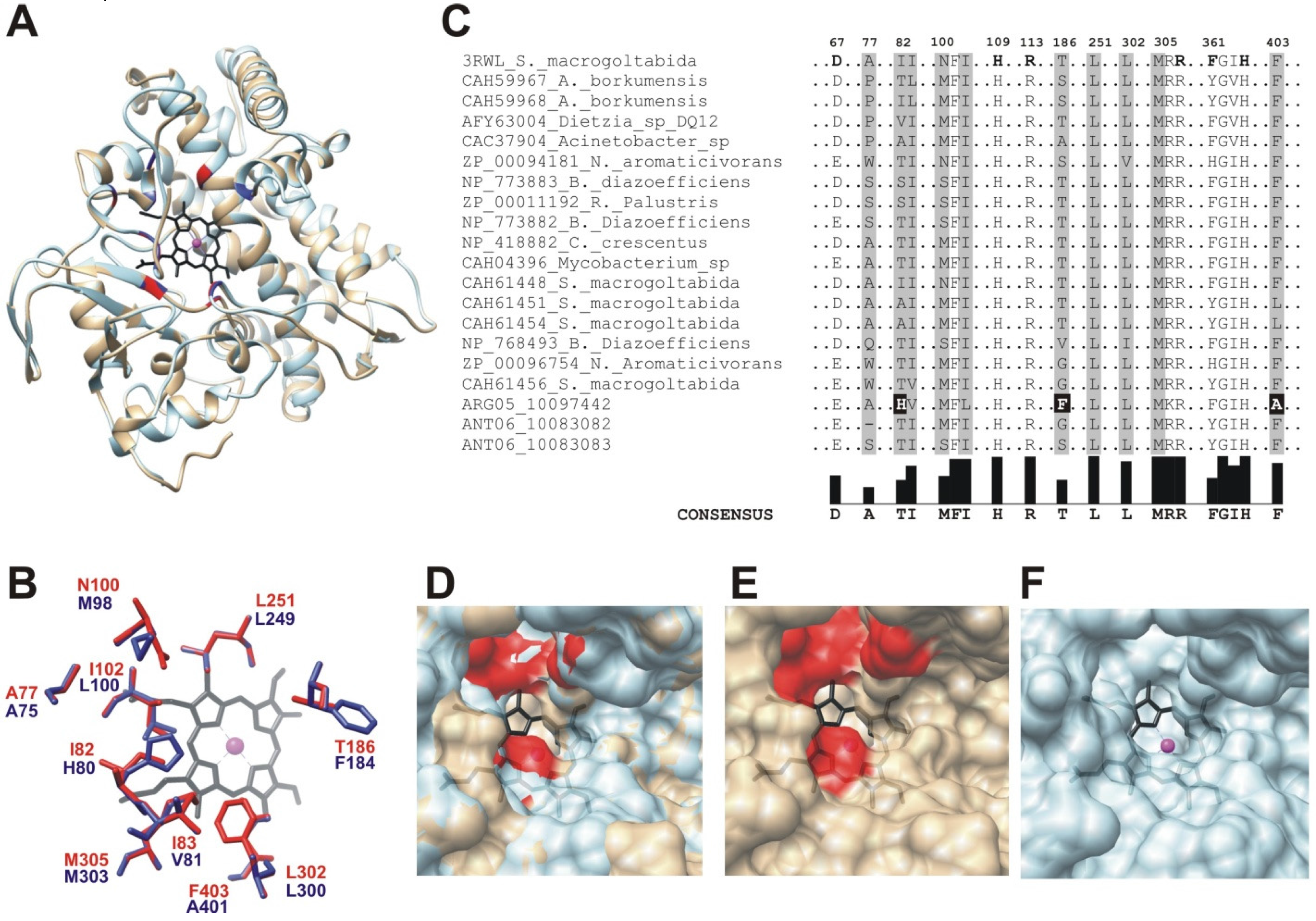
2.2.1. Structural Modeling
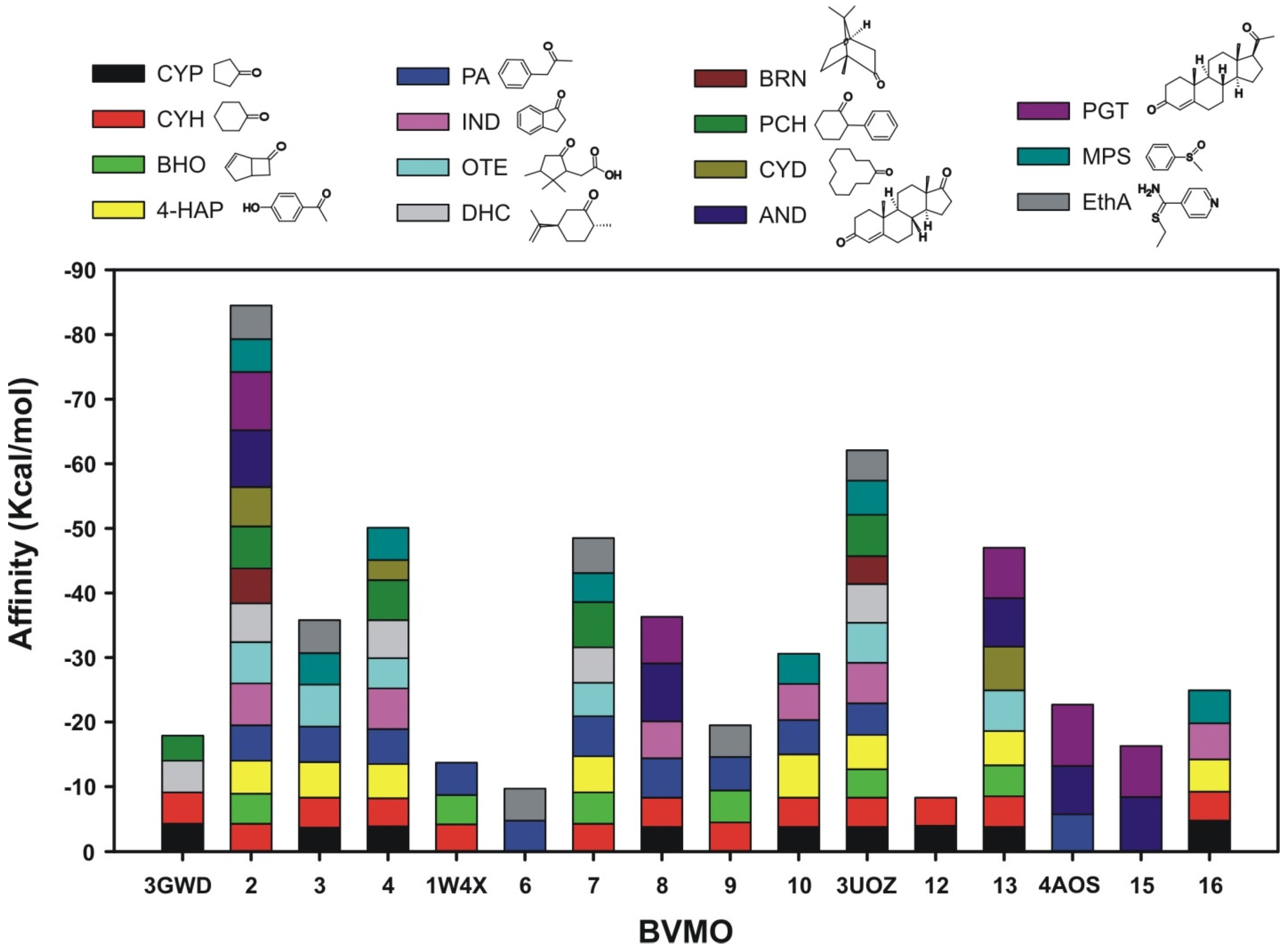
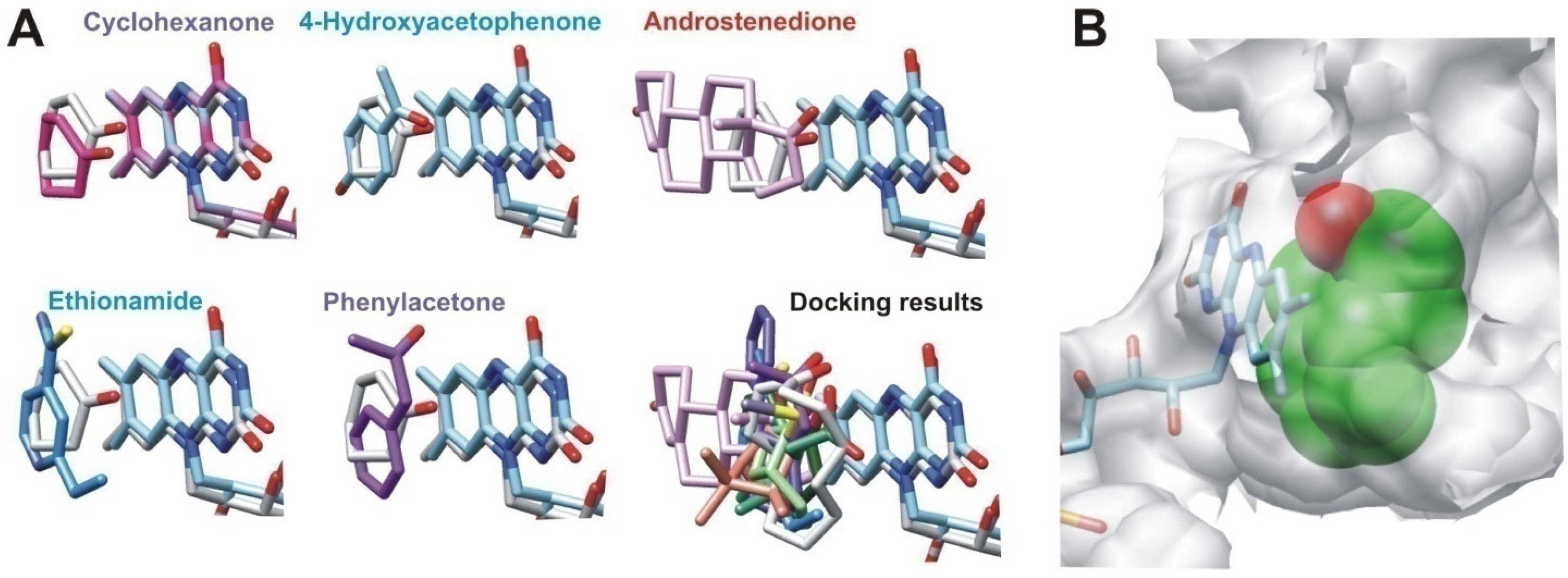
2.2.2. Structural Analysis of the Identified BVMOs: Substrate Range
2.2.3. Structural Flexibility
2.2.4. Structural Analysis of Identified BVMOs: Substrate Regioselectivity
2.3. Cytochrome P450 CYP153
3. Materials and Methods
3.1. Characteristic of Cold Sediments’ Metagenomic Dataset
3.2. Screening of the Metagenomic Dataset
3.3. Phylogenetic Analysis
3.4. Three-Dimensional Protein Structure Modeling and Model Quality Evaluation
3.5. Docking Analysis
3.6. Calculation of Structural Parameters
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Trincone, A. Some enzymes in marine environment: Prospective applications found in patent literature. Recent Patents Biotechnol. 2012, 6, 134–148. [Google Scholar] [CrossRef]
- Lozada, M.; Dionisi, H.M. Microbial bioprospecting in marine environments. In Springer Handbook of Marine Biotechnology; Kim, S.K., Ed.; Springer: Berlin, Germany, 2015; pp. 307–326. [Google Scholar]
- Delmont, T.O.; Simonet, P.; Vogel, T.M. Describing microbial communities and performing global comparisons in the omic era. ISME J.-Int. Soc. Microb. Ecol. 2012, 6, 1625–1628. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.J.; Coleman, N.V. Evolutionary ecology and multidisciplinary approaches to prospecting for monooxygenases as biocatalysts. Antonie Leeuwenhoek 2008, 94, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Martinez-Martinez, M.; Bargiela, R.; Streit, W.R.; Golyshina, O.V.; Golyshin, P.N. Estimating the success of enzyme bioprospecting through metagenomics: Current status and future trends. Microb. Biotechnol. 2015, 9, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Currin, A.; Swainston, N.; Day, P.J.; Kell, D.B. Synthetic biology for the directed evolution of protein biocatalysts: Navigating sequence space intelligently. Chem. Soc. Rev. 2015, 44, 1172–1239. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, D.; Fraaije, M.W.; Arends, I.W.; Opperman, D.J.; Hollmann, F. The taming of oxygen: Biocatalytic oxyfunctionalisations. Chem. Commun. 2014, 50, 13180–13200. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Ding, Y. Recent advances in biocatalyst discovery, development and applications. Bioorg. Med. Chem. 2014, 22, 5604–5612. [Google Scholar] [CrossRef] [PubMed]
- Pazmino, D.T.; Winkler, M.; Glieder, A.; Fraaije, M. Monooxygenases as biocatalysts: Classification, mechanistic aspects and biotechnological applications. J. Biotechnol. 2010, 146, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; van Beilen, J.B.; Duetz, W.A.; Schmid, A.; de Raadt, A.; Griengl, H.; Witholt, B. Oxidative biotransformations using oxygenases. Curr. Opin. Chem. Biol. 2002, 6, 136–144. [Google Scholar] [CrossRef]
- Bučko, M.; Gemeiner, P.; Schenkmayerová, A.; Krajčovič, T.; Rudroff, F.; Mihovilovič, M.D. Baeyer-villiger oxidations: Biotechnological approach. Appl. Microbiol. Biotechnol. 2016, 100, 6585–6599. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, R.; Urlacher, V.B. Cytochromes p450 as promising catalysts for biotechnological application: Chances and limitations. Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Mao, G.; Wang, Y.; Bartlam, M. Structural insights into diversity and n-alkane biodegradation mechanisms of alkane hydroxylases. Front. Microbiol. 2013, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, M.M.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Ceccoli, R.D.; Bianchi, D.A.; Rial, D.V. Flavoprotein monooxygenases for oxidative biocatalysis: Recombinant expression in microbial hosts and applications. Recomb. Protein Expr. Microb. Syst. 2014, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Alphand, V.; Wohlgemuth, R. Applications of baeyer-villiger monooxygenases in organic synthesis. Curr. Org. Chem. 2010, 14, 1928–1965. [Google Scholar] [CrossRef]
- Fraaije, M.W.; Janssen, D.B. Biocatalytic scope of baeyer-villiger monooxygenases. In Modern Biooxidation: Enzymes, reaction and applications; Schmid, R.D., Urlacher, V.B., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA.: Darmstadt, Germany, 2007; pp. 77–97. [Google Scholar]
- Bong, Y.K.; Collier, S.J.; Mijts, B.; Vogel, M.; Zhang, X.; Zhu, J.; Nazor, J.S.D.; Song, S. Synthesis of Prazole Compounds. WO2011071982 A2, 2011. [Google Scholar]
- Leisch, H.; Morley, K.; Lau, P.C. Baeyer-villiger monooxygenases: More than just green chemistry. Chem. Rev. 2011, 111, 4165–4222. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Chi, C.-Q.; Fang, H.; Liang, J.-L.; Lu, S.-L.; Lai, G.-L.; Tang, Y.-Q.; Wu, X.-L. Diverse alkane hydroxylase genes in microorganisms and environments. Sci. Rep. 2014, 4, 4968. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.N.; Lozada, M.; Anselmino, L.E.; Musumeci, M.A.; Henrissat, B.; Jansson, J.K.; Mac Cormack, W.P.; Carroll, J.; Sjöling, S.; Lundgren, L. Metagenomics unveils the attributes of the alginolytic guilds of sediments from four distant cold coastal environments. Environ. Microbiol. 2016, 18, 4471–4484. [Google Scholar] [CrossRef] [PubMed]
- Raddadi, N.; Cherif, A.; Daffonchio, D.; Neifar, M.; Fava, F. Biotechnological applications of extremophiles, extremozymes and extremolytes. Appl. Microbiol. Biotechnol. 2015, 99, 7907–7913. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.C.; Jansson, J.K.; Malfatti, S.A.; Tringe, S.G.; Tiedje, J.M.; Brown, C.T. Tackling soil diversity with the assembly of large, complex metagenomes. Proc. Natl. Acad. Sci. USA 2014, 111, 4904–4909. [Google Scholar] [CrossRef] [PubMed]
- Mascotti, M.L.; Lapadula, W.J.; Ayub, M.J. The origin and evolution of baeyer-villiger monooxygenases (bvmos): An ancestral family of flavin monooxygenases. PLoS ONE 2015, 10, e0132689. [Google Scholar] [CrossRef] [PubMed]
- Fraaije, M.W.; Kamerbeek, N.M.; van Berkel, W.J.; Janssen, D.B. Identification of a baeyer-villiger monooxygenase sequence motif. FEBS Lett. 2002, 518, 43–47. [Google Scholar] [CrossRef]
- Riebel, A.; Dudek, H.; De Gonzalo, G.; Stepniak, P.; Rychlewski, L.; Fraaije, M. Expanding the set of rhodococcal baeyer-villiger monooxygenases by high-throughput cloning, expression and substrate screening. Appl. Microbiol. Biotechnol. 2012, 95, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Yachnin, B.J.; Lau, P.C.; Berghuis, A.M. The role of conformational flexibility in baeyer-villiger monooxygenase catalysis and structure. Biochim. Biophys. Acta 2016, 1864, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Balke, K.; Schmidt, S.; Genz, M.; Bornscheuer, U.T. Switching the regioselectivity of a cyclohexanone monooxygenase toward (+)-trans-dihydrocarvone by rational protein design. ACS Chem. Biol. 2016, 11, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Dudek, H.M.; de Gonzalo, G.; Pazmiño, D.E.T.; Stępniak, P.; Wyrwicz, L.S.; Rychlewski, L.; Fraaije, M.W. Mapping the substrate binding site of phenylacetone monooxygenase from thermobifida fusca by mutational analysis. Appl. Environ. Microbiol. 2011, 77, 5730–5738. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, S.; van Beek, H.L.; Pennetta, A.; Martinoli, C.; Fraaije, M.W.; Mattevi, A. Exploring the structural basis of substrate preferences in baeyer-villiger monooxygenases insight from steroid monooxygenase. J. Biol. Chem. 2012, 287, 22626–22634. [Google Scholar] [CrossRef] [PubMed]
- Yachnin, B.J.; Sprules, T.; McEvoy, M.B.; Lau, P.C.; Berghuis, A.M. The substrate-bound crystal structure of a baeyer-villiger monooxygenase exhibits a criegee-like conformation. J. Am. Chem. Soc. 2012, 134, 7788–7795. [Google Scholar] [CrossRef] [PubMed]
- Mirza, I.A.; Yachnin, B.J.; Wang, S.; Grosse, S.; Bergeron, H.; Imura, A.; Iwaki, H.; Hasegawa, Y.; Lau, P.C.; Berghuis, A.M. Crystal structures of cyclohexanone monooxygenase reveal complex domain movements and a sliding cofactor. J. Am. Chem. Soc. 2009, 131, 8848–8854. [Google Scholar] [CrossRef] [PubMed]
- Leisch, H.; Shi, R.; Grosse, S.; Morley, K.; Bergeron, H.; Cygler, M.; Iwaki, H.; Hasegawa, Y.; Lau, P.C. Cloning, baeyer-villiger biooxidations, and structures of the camphor pathway 2-oxo-δ3-4, 5, 5-trimethylcyclopentenylacetyl-coenzyme a monooxygenase of pseudomonas putida atcc 17453. Appl. Environ. Microbiol. 2012, 78, 2200–2212. [Google Scholar] [CrossRef] [PubMed]
- Malito, E.; Alfieri, A.; Fraaije, M.W.; Mattevi, A. Crystal structure of a baeyer-villiger monooxygenase. Proc. Natl. Acad. Sci. USA 2004, 101, 13157–13162. [Google Scholar] [CrossRef] [PubMed]
- Voss, N.R.; Gerstein, M. 3v: Cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010, 38, W555–W562. [Google Scholar] [CrossRef] [PubMed]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. Castp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Orru, R.; Dudek, H.M.; Martinoli, C.; Pazmiño, D.E.T.; Royant, A.; Weik, M.; Fraaije, M.W.; Mattevi, A. Snapshots of enzymatic baeyer-villiger catalysis oxygen activation and intermediate stabilization. J. Biol. Chem. 2011, 286, 29284–29291. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Feller, G.; Gerday, C. Psychrophilic enzymes: Hot topics in cold adaptation. Nat. Rev. Microbiol. 2003, 1, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Yachnin, B.J.; McEvoy, M.B.; MacCuish, R.J.; Morley, K.L.; Lau, P.C.; Berghuis, A.M. Lactone-bound structures of cyclohexanone monooxygenase provide insight into the stereochemistry of catalysis. ACS Chem. Biol. 2014, 9, 2843–2851. [Google Scholar] [CrossRef] [PubMed]
- Černuchová, P.; Mihovilovic, M.D. Microbial baeyer-villiger oxidation of terpenones by recombinant whole-cell biocatalysts—Formation of enantiocomplementary regioisomeric lactones. Org. Biomol. Chem. 2007, 5, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Van Beilen, J.; Funhoff, E.; Van Loon, A.; Just, A.; Kaysser, L.; Bouza, M.; Holtackers, R.; Rothlisberger, M.; Li, Z.; Witholt, B. Cytochrome P450 alkane hydroxylases of the CYP153 family are common in alkane-degrading eubacteria lacking integral membrane alkane hydroxylases. Appl. Environ. Microbiol. 2006, 72, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Pham, S.Q.; Pompidor, G.; Liu, J.; Li, X.-D.; Li, Z. Evolving P450pyr hydroxylase for highly enantioselective hydroxylation at non-activated carbon atom. Chem. Commun. 2012, 48, 4618–4620. [Google Scholar] [CrossRef] [PubMed]
- Taneja, S.C.; Aga, M.A.; Kumar, B.; Sethi, V.K.; Andotra, S.S.; Qazi, G.N. Process for the Preparation of Optically Active N-Benzyl-3 Hydroxypyrrolidines. US8445700 B2, 2012. [Google Scholar]
- Yang, Y.; Li, Z. Evolving P450pyr monooxygenase for regio-and stereoselective hydroxylations. Chim. Int. J. Chem. 2015, 69, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Chen, I.-M.A.; Chu, K.; Szeto, E.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Pagani, I.; Tringe, S. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 2014, 42, D568–D573. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.L.; Kelly, D.E. Microbial cytochromes p450: Biodiversity and biotechnology. Where do cytochromes p450 come from, what do they do and what can they do for us? Phil. Trans. R. Soc. B 2013, 368. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using modeller. Curr. Protoc. Bioinform. 2006, 47, 5. [Google Scholar]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. Verify3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396. [Google Scholar] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Cheng, J. 3Drefine: Consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins 2013, 81, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. Swiss-model and the swiss-pdb viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. Pubchem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | B-FDM a | CYP153 b | Identified Sequences/Metagenome | PCS c | Assembly (%) d |
|---|---|---|---|---|---|
| NOR02 | 0 | 0 | 0 | 2.82 × 105 | 12.99 |
| NOR05 | 8 | 3 | 11 | 7.85 × 105 | 22.74 |
| NOR08 | 11 | 15 | 26 | 1.28 × 106 | 29.04 |
| NOR13 | 2 | 1 | 3 | 3.13 × 105 | 17.00 |
| NOR15 | 9 | 7 | 16 | 1.47 × 106 | 34.55 |
| NOR18 | 3 | 1 | 4 | 3.92 × 105 | 23.55 |
| SWE02 | 2 | 5 | 7 | 1.11 × 106 | 19.29 |
| SWE07 | 0 | 0 | 0 | 2.68 × 105 | 8.77 |
| SWE12 | 7 | 1 | 8 | 1.08 × 106 | 18.17 |
| SWE21 | 6 | 3 | 9 | 8.66 × 105 | 17.10 |
| SWE26 | 0 | 0 | 0 | 5.39 × 105 | 12.03 |
| ARG01 | 0 | 1 | 1 | 1.39 × 105 | 4.50 |
| ARG02 | 0 | 1 | 1 | 1.74 × 105 | 5.20 |
| ARG03 | 0 | 0 | 0 | 4.74 × 105 | 13.86 |
| ARG04 | 1 | 3 | 4 | 2.79 × 105 | 8.15 |
| ARG05 | 6 | 8 | 14 | 7.13 × 105 | 13.81 |
| ARG06 | 0 | 0 | 0 | 1.87 × 105 | 5.21 |
| ANT01 | 19 | 32 | 51 | 1.11 × 106 | 29.19 |
| ANT02 | 10 | 23 | 33 | 9.72 × 105 | 27.56 |
| ANT03 | 1 | 4 | 5 | 2.79 × 105 | 13.90 |
| ANT04 | 6 | 19 | 25 | 6.57 × 105 | 26.45 |
| ANT05 | 6 | 12 | 18 | 4.67 × 105 | 20.40 |
| ANT06 | 11 | 17 | 28 | 1.01 × 105 | 7.80 |
| Total | 108 | 156 | 264 | 1.39 × 107 |
| Enzyme | ΔGint (a) (kcal/mol) | ΔGdiss (b) (kcal/mol) | NHB (c) | NSB (d) |
|---|---|---|---|---|
| 3GWD | −30.8 | 35.2 | 38 | 6 |
| NOR08_100243532 | −26.5 | 22.6 | 21 | 0 |
| ANT05_100010021 | −22.1 | 17.7 | 18 | 5 |
| NOR08_100070122 | −28.1 | 23.5 | 18 | 3 |
| 1W4X | −51.3 | 28.3 | 49 | 5 |
| ANT01_100026088 | −17.7 | 10.7 | 13 | 4 |
| SWE21_100067072 | −24.1 | 18.3 | 22 | 7 |
| 3UOZ | −30.5 | 33.2 | 35 | 4 |
| Structure of Lactone | BVMO | |||
|---|---|---|---|---|
| Products | ANT05_100010021 | NOR08_100243532 | NOR08_100070122 | 4RG3 |
A-  | −4.5 | -4.7 | -5.0 | -6.0 |
B- (Normal) | −5.3 | NB | NB | NB |
C- (Abnormal) | −5.1 | -5.0 | -5.5 | -6.2 |
D- (Normal) | −6.1 | NB | NB | NB |
E- (Abnormal)  | −5.8 | NB | -5.9 | -4.5 |
F- (Normal) | −6.9 | NB | NB | NB |
G- (Abnormal) | −4.9 | NB | -6.2 | NB |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musumeci, M.A.; Lozada, M.; Rial, D.V.; Mac Cormack, W.P.; Jansson, J.K.; Sjöling, S.; Carroll, J.; Dionisi, H.M. Prospecting Biotechnologically-Relevant Monooxygenases from Cold Sediment Metagenomes: An In Silico Approach. Mar. Drugs 2017, 15, 114. https://doi.org/10.3390/md15040114
Musumeci MA, Lozada M, Rial DV, Mac Cormack WP, Jansson JK, Sjöling S, Carroll J, Dionisi HM. Prospecting Biotechnologically-Relevant Monooxygenases from Cold Sediment Metagenomes: An In Silico Approach. Marine Drugs. 2017; 15(4):114. https://doi.org/10.3390/md15040114
Chicago/Turabian StyleMusumeci, Matías A., Mariana Lozada, Daniela V. Rial, Walter P. Mac Cormack, Janet K. Jansson, Sara Sjöling, JoLynn Carroll, and Hebe M. Dionisi. 2017. "Prospecting Biotechnologically-Relevant Monooxygenases from Cold Sediment Metagenomes: An In Silico Approach" Marine Drugs 15, no. 4: 114. https://doi.org/10.3390/md15040114
APA StyleMusumeci, M. A., Lozada, M., Rial, D. V., Mac Cormack, W. P., Jansson, J. K., Sjöling, S., Carroll, J., & Dionisi, H. M. (2017). Prospecting Biotechnologically-Relevant Monooxygenases from Cold Sediment Metagenomes: An In Silico Approach. Marine Drugs, 15(4), 114. https://doi.org/10.3390/md15040114