Mertensene, a Halogenated Monoterpene, Induces G2/M Cell Cycle Arrest and Caspase Dependent Apoptosis of Human Colon Adenocarcinoma HT29 Cell Line through the Modulation of ERK-1/-2, AKT and NF-κB Signaling


 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
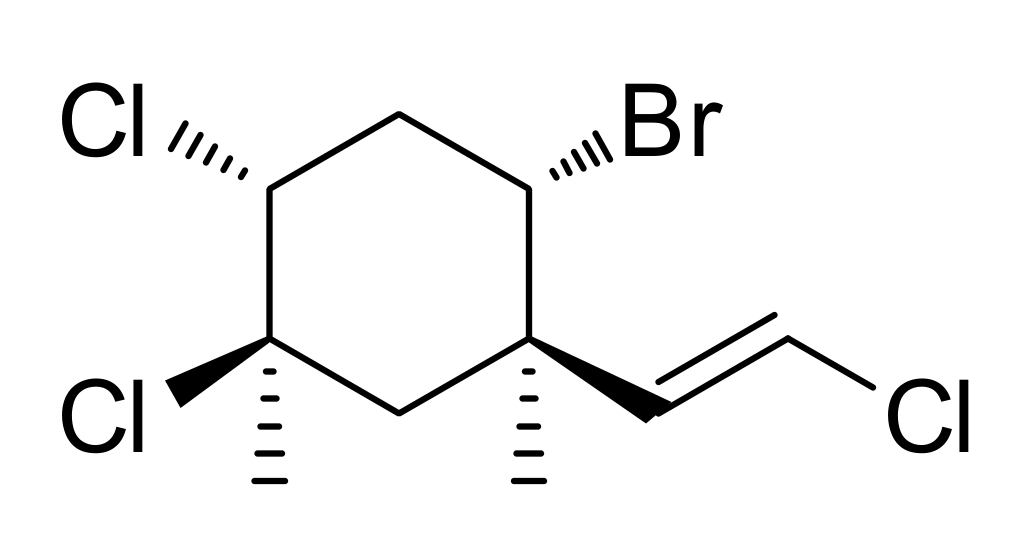
2.1. Isolation and Identification of Mertensene
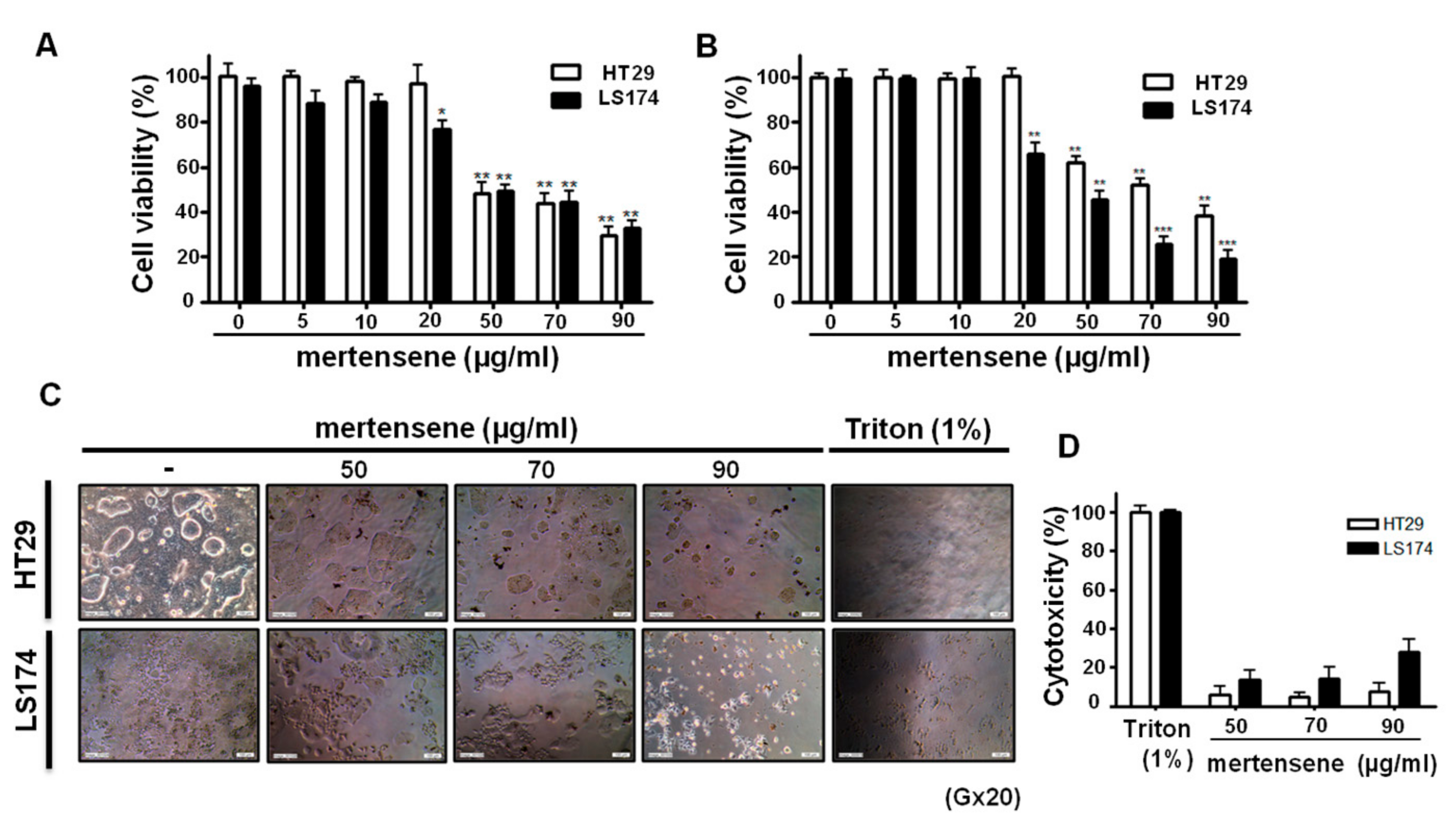
2.2. Mertensene Affects the Viability of HT29 and LS174 Human Colon Adenocarcinoma Cells Independently of Their p53 Status
2.3. Mertensene Did Not Induce Plasmatic Membrane Damage
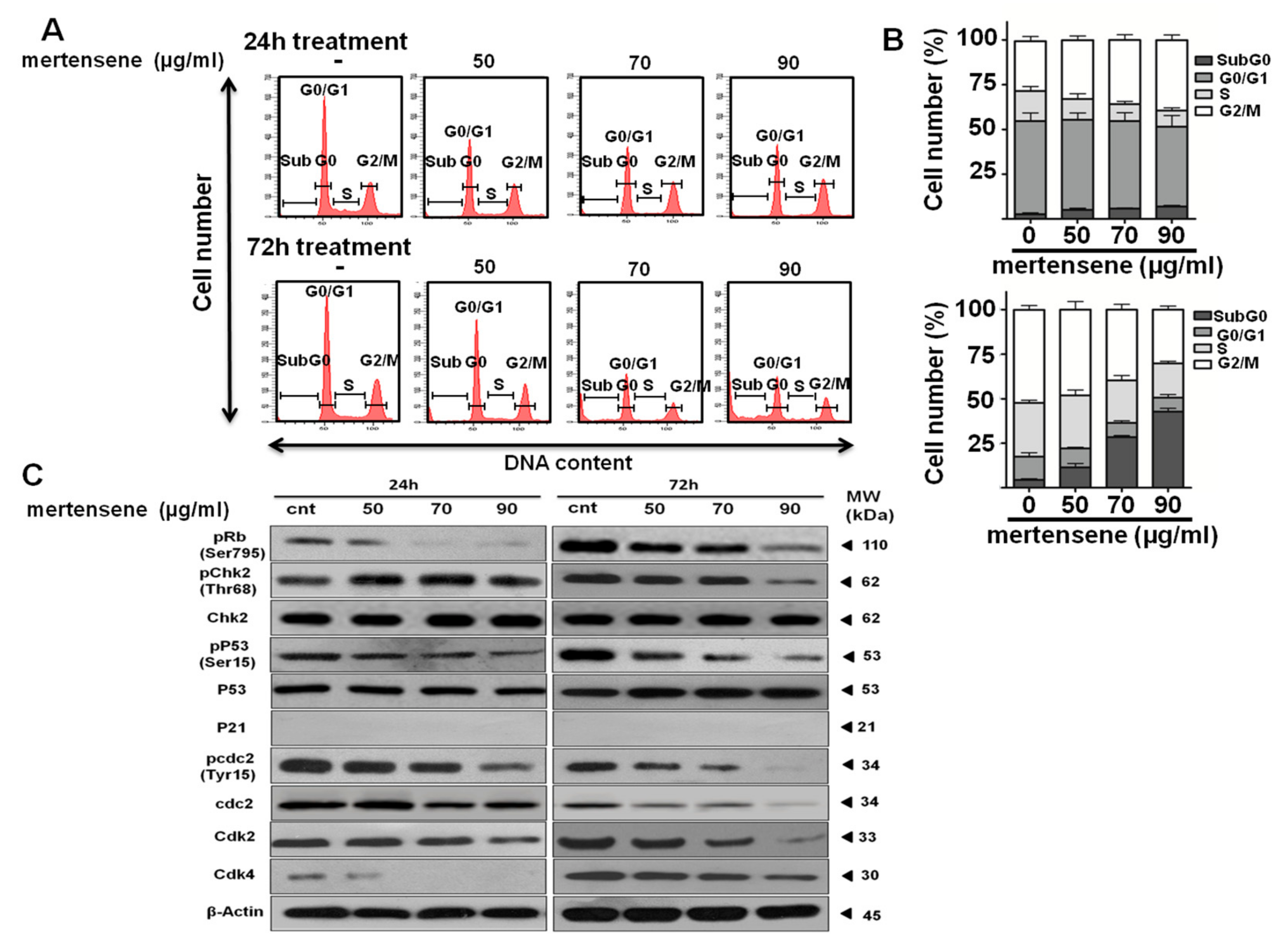
2.4. Mertensene-Induced G2/M Cell Cycle Arrest Is Mediated by Related Regulatory Effectors
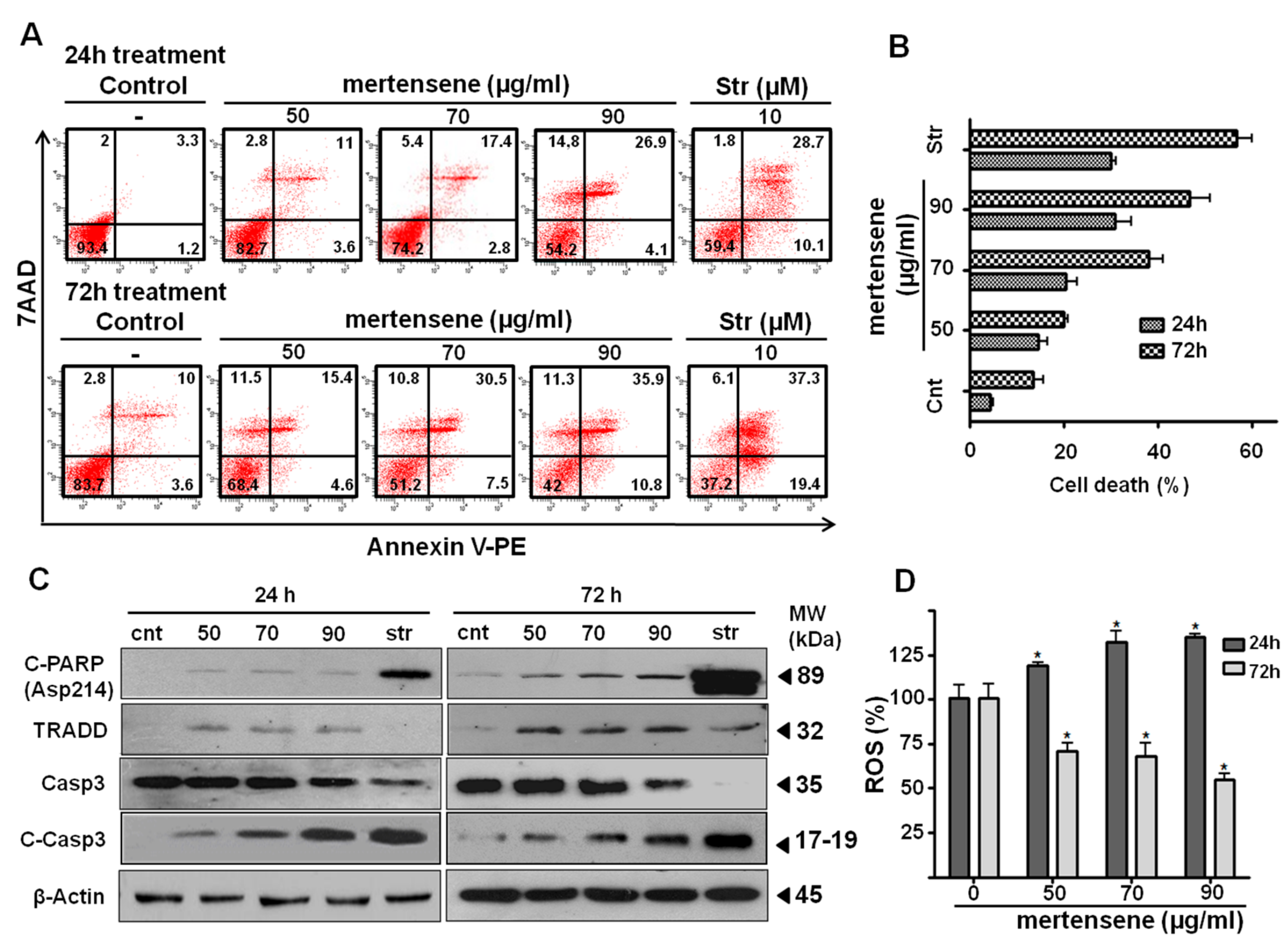
2.5. Mertensene Induces Apoptotic Cell Death in HT29 Cells
2.6. Mertensene Modulates ROS Production
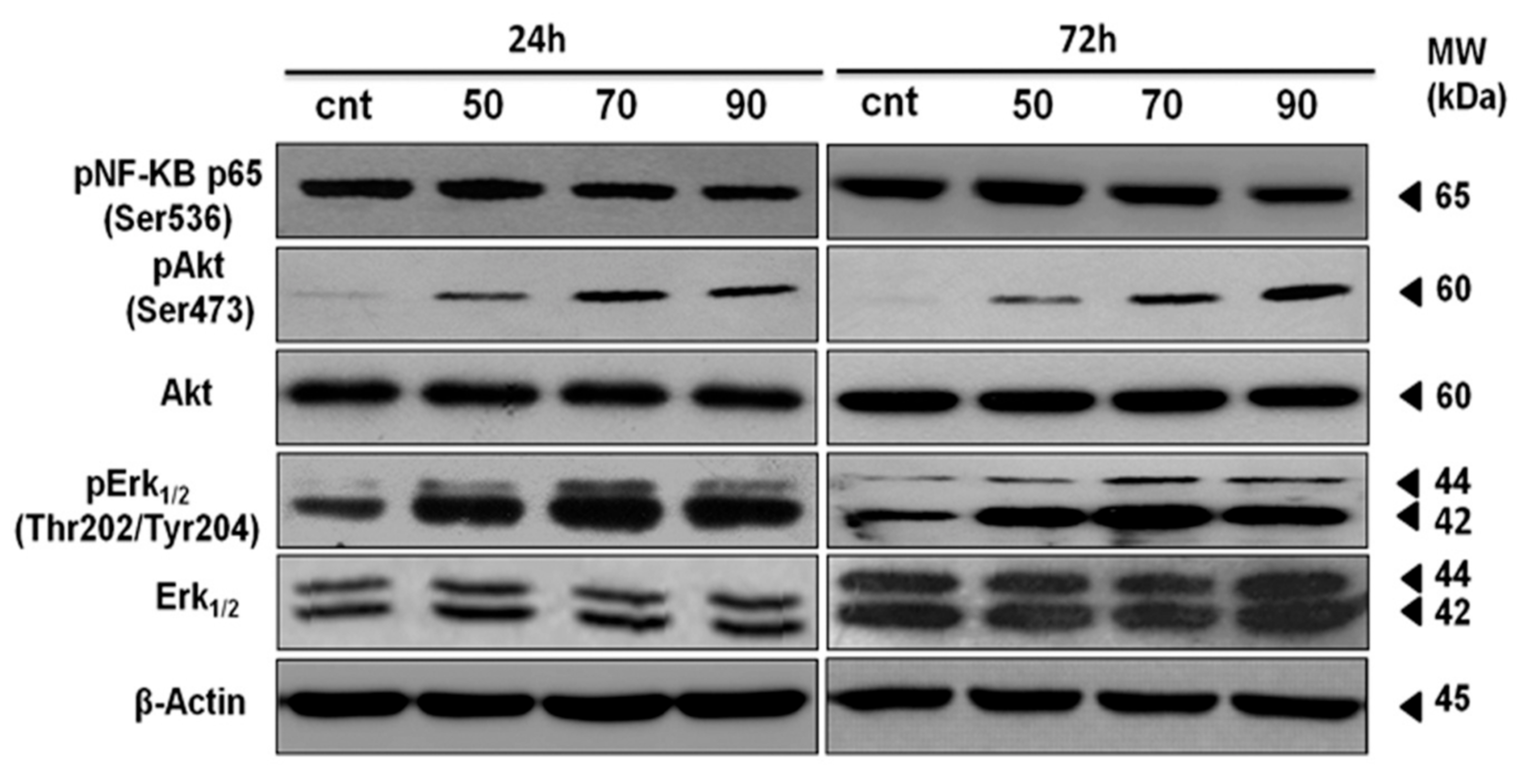
2.7. ERK-1/-2, AKT and NF-κB Activation Contributes to Mertensene-Induced Inhibition of HT29 Cell Viability
3. Discussion
4. Experimental Section
4.1. Extraction and Isolation of Mertensene
4.2. Chemical Studies
4.3. Pharmacological Studies
4.3.1. Cell Cultures
4.3.2. Cell Viability Assays
4.3.3. LDH Cytotoxicity Assay
4.3.4. Cell Cycle Analysis
4.3.5. Cell Apoptosis Analysis
4.3.6. Detection of Intracellulaar Reactive Oxygen Species (ROS)
4.3.7. Western Blot Analysis
4.4. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kuttan, G.; Pratheeshkumar, P.; Manu, K.A.; Kuttan, R. Inhibition of tumor progression by naturally occurring terpenoids. Pharm. Biol. 2011, 49, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Abhishek Bhanot, R.S.; Malleshappa, N.N. Natural sources as potential anti-cancer agents: A review. Phytomedicine 2011, 3, 9–26. [Google Scholar]
- Hussain, S.M.; Fareed, S.; Ansari, S.; Saba, M.S. Marine natural products: A lead for anti-cancer. Indian J. Mar. Sci. 2012, 41, 27–39. [Google Scholar]
- Goulitquer, S.; Potin, P.; Tonon, T. Mass spectrometry-based metabolomics to elucidate functions in marine organisms and ecosystems. Mar. Drugs 2012, 10, 849–880. [Google Scholar] [CrossRef] [PubMed]
- Von Schwarzenberg, K.; Vollmar, A.M. Targeting apoptosis pathways by natural compounds in cancer: Marine compounds as lead structures and chemical tools for cancer therapy. Cancer Lett. 2013, 332, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Schwartsmann, G.; da Rocha, A.B.; Berlinck, R.G.; Jimeno, J. Marine organisms as a source of new anticancer agents. Lancet Oncol. 2001, 2, 221–225. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Hu, W.P.; Munro, M.H.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2009, 26, 170–244. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, E.; Roussis, V. Natural products from seaweeds. In Plant-Derived Natural Products; Osbourn, A.E., Lanzotti, V., Eds.; Springer: New York, NY, USA, 2009; pp. 51–81. [Google Scholar]
- Moussavou, G.; Kwak, D.H.; Obiang-Obonou, B.W.; Ogandaga Maranguy, C.A.; Dinzouna-Boutamba, S.D.; Lee, D.H.; Pissibanganga, O.G.M.; Ko, K.; Seo, J.I.; Choo, Y.K. Anticancer effects of different seaweeds on human colon and breast cancers. Mar. Drugs 2014, 12, 4898–4911. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.C.; Costa-Lotufo, L.V. Bioprospecting for bioactives from seaweeds: Potential, obstacles and alternatives. Rev. Bras. Farm. 2012, 22, 894–905. [Google Scholar] [CrossRef]
- Eric, H.; Andrianasolo, D.F.; Cornell-Kennon, S.; William, H.G. DNA methyl transferase inhibiting halogenated monoterpenes from the madagascar red marine alga Portieria hornemannii. J. Nat. Prod. 2006, 69, 576–579. [Google Scholar]
- De Ines, C.; Argandona, V.H.; Rovirosa, J.; San-Martin, A.; Diaz-Marrero, A.R.; Cueto, M.; Gonzalez-Coloma, A. Cytotoxic activity of halogenated monoterpenes from Plocamium cartilagineum. Z. Naturforsch. 2004, 59, 339–344. [Google Scholar]
- Antunes, E.M.; Afolayan, A.F.; Chiwakata, M.T.; Fakee, J.; Knott, M.G.; Whibley, C.E.; Hendricks, D.T.; Bolton, J.J.; Beukes, D.R. Identification and in vitro anti-esophageal cancer activity of a series of halogenated monoterpenes isolated from the south african seaweeds Plocamium suhrii and Plocamium cornutum. Phytochemistry 2011, 72, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Knott, M.G.; Mkwananzi, H.; Arendse, C.E.; Hendricks, D.T.; Bolton, J.J.; Beukes, D.R. Plocoralides a–c, polyhalogenated monoterpenes from the marine alga Plocamium corallorhiza. Phytochemistry 2005, 66, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.G.A.; Mkwananzi, H.B.; Antunes, E.M.; Whibley, C.E.; Hendricks, D.T.; Bolton, J.J.; Beukes, D.R. Halogenated monoterpene aldehydes from the south african marine alga Plocamium corallorhiza. J. Nat. Prod. 2007, 70, 596–599. [Google Scholar] [CrossRef] [PubMed]
- De la Mare, J.-A.; Lawson, J.; Chiwakata, M.; Beukes, D.; Edkins, A.; Blatch, G. Quinones and halogenated monoterpenes of algal origin show anti-proliferative effects against breast cancer cells in vitro. Investig. New Drugs 2012, 30, 2187–2200. [Google Scholar] [CrossRef] [PubMed]
- Capon, R.; Engelhardt, L.; Ghisalberti, E.; Jefferies, P.; Patrick, V.; White, A. Structural studies of polyhalogenated monoterpenes from Plocamium species. Aust. J. Chem. 1984, 37, 537–544. [Google Scholar] [CrossRef]
- Errea, M.I.; Matulewicz, M.C. The international journal of plant biochemistrycold water-soluble polysaccharides from tetrasporic Pterocladia capillacea. Phytochemistry 1994, 37, 1075–1078. [Google Scholar] [CrossRef]
- Errea, M.I.; Matulewicz, M.C. Unusual structures in the polysaccharides from the red seaweed Pterocladiella capillacea (Gelidiaceae, Gelidiales). Carbohydr. Res. 2003, 338, 943–953. [Google Scholar] [CrossRef]
- Errea, M.I.; Matulewicz, M.C. Hot water-soluble polysaccharides from tetrasporic Pterocladia capillaceae. Phytochemistry 1996, 42, 1071–1073. [Google Scholar] [CrossRef]
- Rao, A.; Bekheet, I.A. Preparation of agar-agar from the red seaweed Pterocladia capillacea off the coast of Alexandria, Egypt. Appl. Environ. Microbiol. 1976, 32, 479–482. [Google Scholar] [PubMed]
- Oliveira, S.R.; Nascimento, A.E.; Lima, M.E.; Leite, Y.; Benevides, N.M. Purification and characterisation of a lectin from the red marine alga Pterocladiella capillacea (sg gmel.) santel. & hommers. Rev. Brasil. Bot. 2002, 25, 397–403. [Google Scholar]
- Whitfield, F.B.; Helidoniotis, F.; Shaw, K.J.; Svoronos, D. Distribution of bromophenols in species of marine algae from eastern Australia. J. Agric. Food Chem. 1999, 47, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Sciuto, S.; Chillemi, R.; Piattelli, M. Onium compounds from the red alga Pterocladia capillacea. J. Nat. Prod. 1988, 51, 322–325. [Google Scholar] [CrossRef]
- Norton, R.S.; Warren, R.G.; Wells, R.J. Three new polyhalogenated monoterpenes from Plocamium species. Tetrahedron Lett. 1977, 18, 3905–3908. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kim, J.; Prasad, S.; Aggarwal, B. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 2010, 29, 405–434. [Google Scholar] [CrossRef] [PubMed]
- Sreevalsan, S.; Safe, S. Reactive oxygen species and colorectal cancer. Curr. Colorectal Cancer Rep. 2013, 9, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, C.R.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C. Roles of the raf/mek/erk and pi3k/pten/akt/mtor pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 2011, 3, 192–222. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. Nf-kb in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Thoppil, R.J.; Bishayee, A. Terpenoids as potential chemopreventive and therapeutic agents in liver cancer. World J. Hepatol. 2011, 3, 228–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, Y. Targeting cancer with sesterterpenoids: The new potential antitumor drugs. J. Nat. Med. 2015, 69, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Rabi, T.; Bishayee, A. Terpenoids and breast cancer chemoprevention. Breast Cancer Res. Treat. 2009, 115, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-Y.; Wang, S.-K.; Ou, Y.-H.; Duh, C.-Y. Mollisolactones a and b, novel dinormonoterpenes from the soft coral Sinularia mollis. Bioorg. Med. Chem. Lett. 2016, 26, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Sabry, O.M.M.; Goeger, D.E.; Valeriote, F.A.; Gerwick, W.H. Cytotoxic halogenated monoterpenes from Plocamium cartilagineum. Nat. Prod. Res. 2017, 31, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Bo, H.; van den Heuvel, A.P.J.; Varun, V.P.; Shengliang, Z.; Wafik, S.E.-D. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar]
- Pavlou, D.; Kirmizis, A. Depletion of histone n-terminal-acetyltransferase naa40 induces p53-independent apoptosis in colorectal cancer cells via the mitochondrial pathway. Apoptosis 2015, 21, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Zhang, H.H.; Zhou, C.X.; Li, C.; Zhang, F.; Mei, Q.B. The histone deacetylase inhibitor trichostatin a induces cell cycle arrest and apoptosis in colorectal cancer cells via p53-dependent and -independent pathways. Oncol. Rep. 2012, 28, 384–388. [Google Scholar] [PubMed]
- Thapa, D.; Babu, D.; Park, M.-A.; Kwak, M.-K.; Lee, Y.-R.; Kim, J.M.; Kwon, T.K.; Kim, J.-A. Induction of p53-independent apoptosis by a novel synthetic hexahydrocannabinol analog is mediated via sp1-dependent nsaid-activated gene-1 in colon cancer cells. Biochem. Pharmacol. 2010, 80, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K.; Shah, M.A. Targeting the cell cycle: A new approach to cancer therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. CA Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef] [PubMed]
- Wong, R. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Koff, J.L.; Ramachandiran, S.; Bernal-Mizrachi, L. A time to kill: Targeting apoptosis in cancer. Int. J. Mol. Sci. 2015, 16, 2942–2955. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.A.; Kirby, R. Apoptosis: A review of pro-apoptotic and anti-apoptotic pathways and dysregulation in disease. J. Vet. Emerg. Crit. Care 2008, 18, 572–585. [Google Scholar] [CrossRef]
- Brenner, D.; Mak, T.W. Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 2009, 21, 871–877. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Janicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Kan, W.L.T.; Yin, C.; Xu, H.X.; Xu, G.; To, K.K.W.; Cho, C.H.; Rudd, J.A.; Lin, G. Antitumor effects of novel compound, guttiferone k, on colon cancer by p21waf1/cip1-mediated g0/g1 cell cycle arrest and apoptosis. Int. J. Cancer 2013, 132, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Weidner, C.; Rousseau, M.; Micikas, R.J.; Fischer, C.; Plauth, A.; Wowro, S.J.; Siems, K.; Hetterling, G.; Kliem, M.; Schroeder, F.C. Amorfrutin c induces apoptosis and inhibits proliferation in colon cancer cells through targeting mitochondria. J. Nat. Prod. 2016, 79, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Madanchi, R.; Hauschild, J.; Otte, K.; Alsdorf, W.H.; Schumacher, U.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Honecker, F.; et al. The marine triterpene glycoside frondoside a induces p53-independent apoptosis and inhibits autophagy in urothelial carcinoma cells. BMC Cancer 2017, 17, 93. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Richardson, B.C. The mapk signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Danielsen, S.A.; Eide, P.W.; Nesbakken, A.; Guren, T.; Leithe, E.; Lothe, R.A. Portrait of the pi3k/akt pathway in colorectal cancer. Biochim. Biophys. Acta 2015, 1855, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Maeda, S. Targeting nf-κb for colorectal cancer. Expert Opin. Ther. Targets 2010, 14, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Maeda, S.; Hikiba, Y.; Nakagawa, H.; Hayakawa, Y.; Shibata, W.; Yanai, A.; Ogura, K.; Omata, M. Constitutive nf-κb activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin. Cancer Res. 2009, 15, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa b to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Los, M.; Walczak, H. Caspases: Their Role in Cell Death and Cell Survival; Molecular Biology Intelligence Unit; Springer: New York, NY, USA, 2002; Volume 24. [Google Scholar]
- Lu, Z.; Xu, S. Erk1/2 map kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Sun, W.; Sun, H.; Wei, S.; Chen, R.; Wang, B.; Huang, C. Asperolide a, a marine-derived tetranorditerpenoid, induces g2/m arrest in human nci-h460 lung carcinoma cells, is mediated by p53-p21 stabilization and modulated by ras/raf/mek/erk signaling pathway. Mar. Drugs 2013, 11, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Hong, Y.; Miao, L.; Li, C.; Xu, G.; Wei, S.; Wang, B.; Huang, C.; Jiao, B. Wentilactone a as a novel potential antitumor agent induces apoptosis and g2/m arrest of human lung carcinoma cells, and is mediated by hras-gtp accumulation to excessively activate the ras/raf/erk/p53-p21 pathway. Cell Death Dis. 2013, 4, e952. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Gandhi, V. Ros-activated anticancer prodrugs: A new strategy for tumor-specific damage. Ther. Deliv. 2012, 3, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarhouni-Jabberi, S.; Zakraoui, O.; Ioannou, E.; Riahi-Chebbi, I.; Haoues, M.; Roussis, V.; Kharrat, R.; Essafi-Benkhadir, K. Mertensene, a Halogenated Monoterpene, Induces G2/M Cell Cycle Arrest and Caspase Dependent Apoptosis of Human Colon Adenocarcinoma HT29 Cell Line through the Modulation of ERK-1/-2, AKT and NF-κB Signaling. Mar. Drugs 2017, 15, 221. https://doi.org/10.3390/md15070221
Tarhouni-Jabberi S, Zakraoui O, Ioannou E, Riahi-Chebbi I, Haoues M, Roussis V, Kharrat R, Essafi-Benkhadir K. Mertensene, a Halogenated Monoterpene, Induces G2/M Cell Cycle Arrest and Caspase Dependent Apoptosis of Human Colon Adenocarcinoma HT29 Cell Line through the Modulation of ERK-1/-2, AKT and NF-κB Signaling. Marine Drugs. 2017; 15(7):221. https://doi.org/10.3390/md15070221
Chicago/Turabian StyleTarhouni-Jabberi, Safa, Ons Zakraoui, Efstathia Ioannou, Ichrak Riahi-Chebbi, Meriam Haoues, Vassilios Roussis, Riadh Kharrat, and Khadija Essafi-Benkhadir. 2017. "Mertensene, a Halogenated Monoterpene, Induces G2/M Cell Cycle Arrest and Caspase Dependent Apoptosis of Human Colon Adenocarcinoma HT29 Cell Line through the Modulation of ERK-1/-2, AKT and NF-κB Signaling" Marine Drugs 15, no. 7: 221. https://doi.org/10.3390/md15070221
APA StyleTarhouni-Jabberi, S., Zakraoui, O., Ioannou, E., Riahi-Chebbi, I., Haoues, M., Roussis, V., Kharrat, R., & Essafi-Benkhadir, K. (2017). Mertensene, a Halogenated Monoterpene, Induces G2/M Cell Cycle Arrest and Caspase Dependent Apoptosis of Human Colon Adenocarcinoma HT29 Cell Line through the Modulation of ERK-1/-2, AKT and NF-κB Signaling. Marine Drugs, 15(7), 221. https://doi.org/10.3390/md15070221