Analgesic Activity of Acid-Sensing Ion Channel 3 (ASIС3) Inhibitors: Sea Anemones Peptides Ugr9-1 and APETx2 versus Low Molecular Weight Compounds

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Whole Cell Electrophysiology
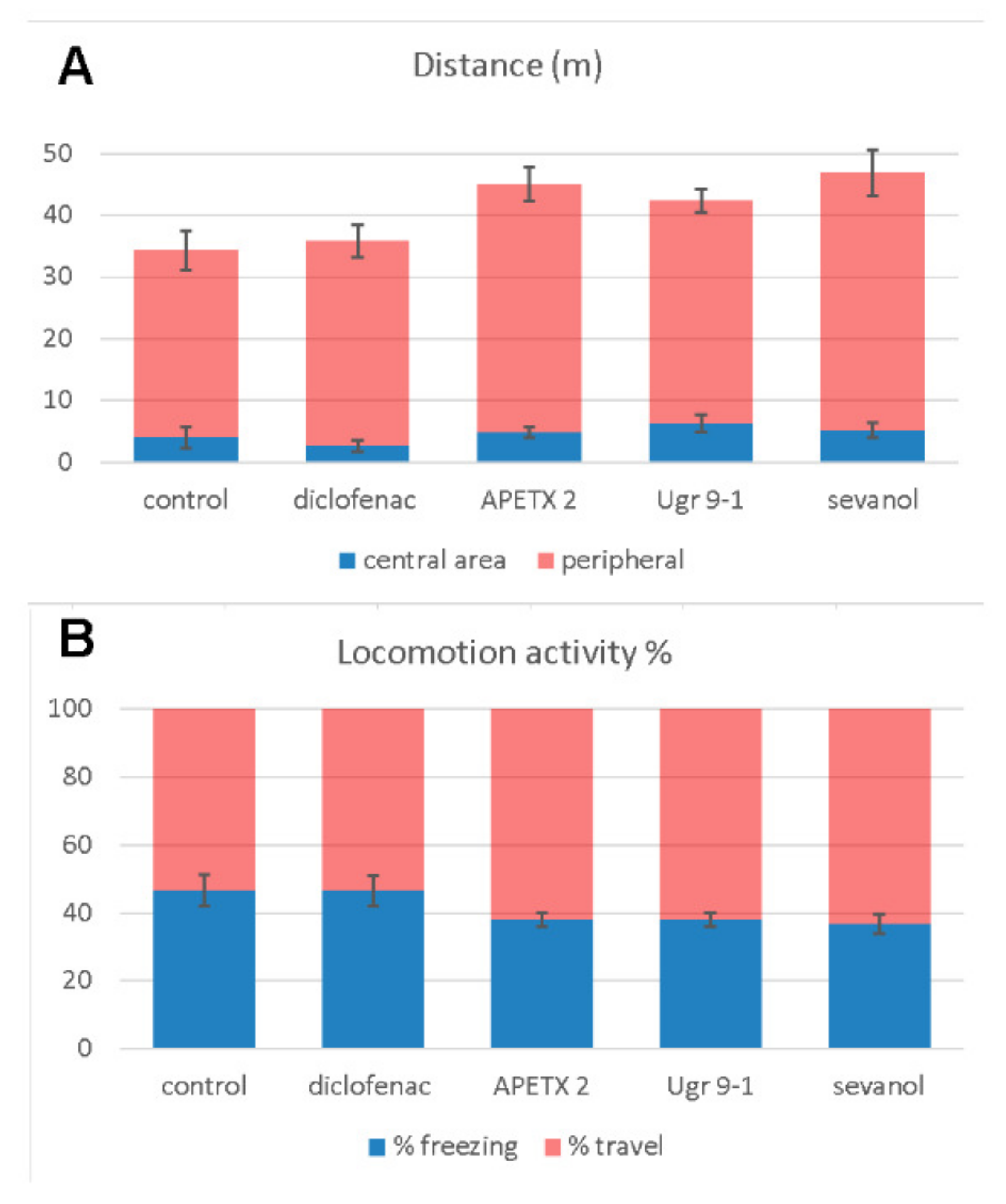
2.2. Open Field Test
2.3. Acetic Acid-Induced Writhing
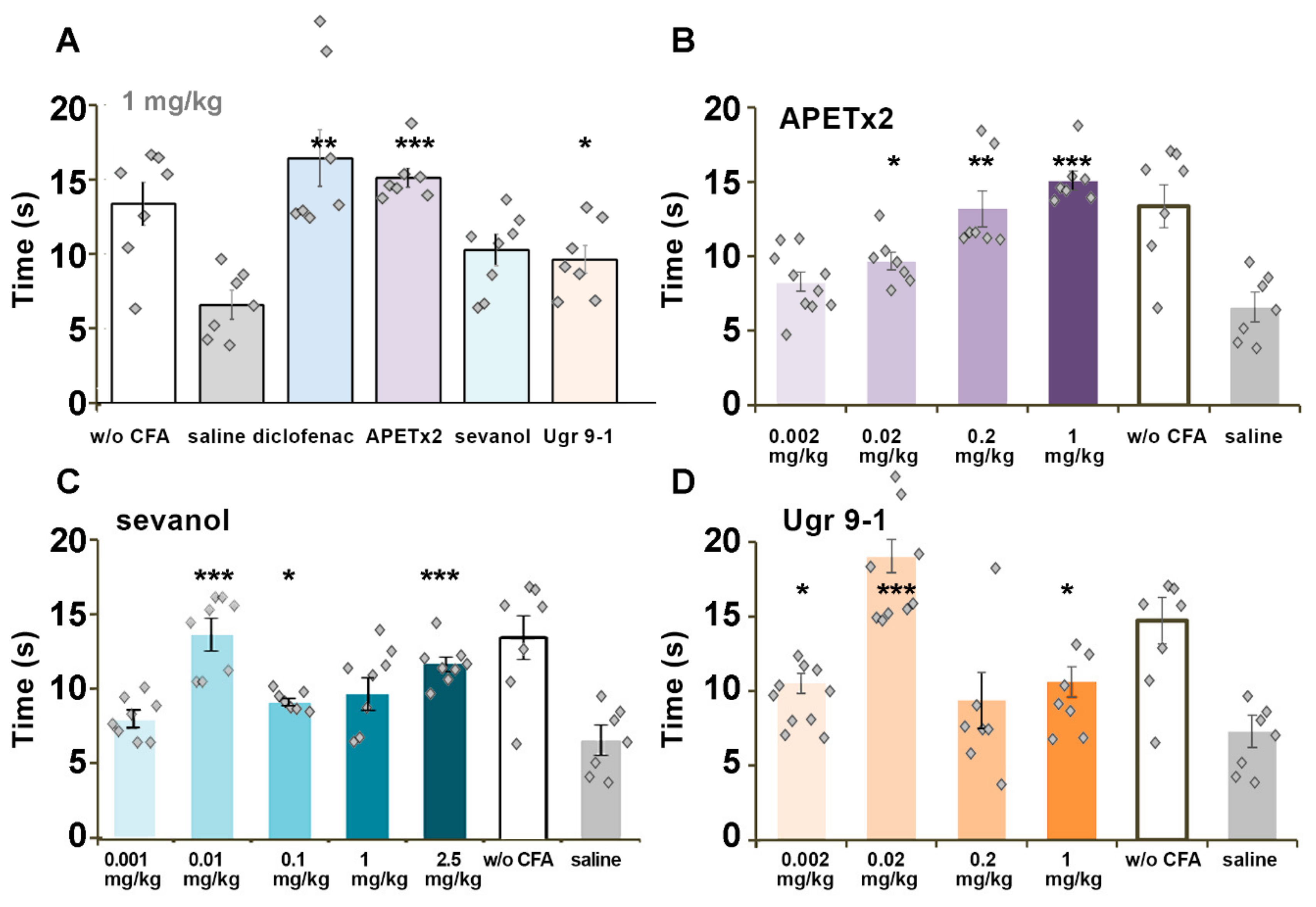
2.4. CFA-Induced Inflammation
3. Discussion
4. Materials and Methods
4.1. Ligands
4.2. Electrophysiology
4.3. Animals
4.3.1. CFA-Induced Thermal Hyperalgesia
4.3.2. Acetic Acid-Induced Writhing (Abdominal Constriction Test of Visceral Pain)
4.3.3. Open Field Test
4.4. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- García-Añoveros, J.; Derfler, B.; Neville-Golden, J.; Hyman, B.T.; Corey, D.P. BNaC1 and BNaC2 constitute a new family of human neuronal sodium channels related to degenerins and epithelial sodium channels. Proc. Natl. Acad. Sci. USA 1997, 94, 1459–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellenberger, S.; Schild, L. International Union of Basic and Clinical Pharmacology. XCI. Structure, Function, and Pharmacology of Acid-Sensing Ion Channels and the Epithelial Na+ Channel. Pharmacol. Rev. 2015, 67, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.-G.; Zhu, X.-M.; Chu, X.-P.; Minami, M.; Hey, J.; Wei, W.-L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Wu, L.-J.; Yu, Y.-Q.; Ding, Y.; Jing, L.; Xu, L.; Chen, J.; Xu, T.-L. Upregulation of acid-sensing ion channel ASIC1a in spinal dorsal horn neurons contributes to inflammatory pain hypersensitivity. J. Neurosci. 2007, 27, 11139–11148. [Google Scholar] [CrossRef] [PubMed]
- Arias, R.L.; Sung, M.-L.A.; Vasylyev, D.; Zhang, M.-Y.; Albinson, K.; Kubek, K.; Kagan, N.; Beyer, C.; Lin, Q.; Dwyer, J.M.; et al. Amiloride is neuroprotective in an MPTP model of Parkinson’s disease. Neurobiol. Dis. 2008, 31, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Komnig, D.; Imgrund, S.; Reich, A.; Gründer, S.; Falkenburger, B.H. ASIC1a Deficient Mice Show Unaltered Neurodegeneration in the Subacute MPTP Model of Parkinson Disease. PLoS ONE 2016, 11, e0165235. [Google Scholar] [CrossRef] [PubMed]
- Deval, E.; Lingueglia, E. Acid-Sensing Ion Channels and nociception in the peripheral and central nervous systems. Neuropharmacology 2015, 94, 49–57. [Google Scholar] [CrossRef]
- Osmakov, D.I.; Andreev, Y.A.; Kozlov, S.A. Acid-sensing ion channels and their modulators. Biochemistry 2014, 79, 1528–1545. [Google Scholar] [CrossRef]
- Babinski, K.; Lê, K.T.; Séguéla, P. Molecular cloning and regional distribution of a human proton receptor subunit with biphasic functional properties. J. Neurochem. 1999, 72, 51–57. [Google Scholar] [CrossRef]
- Price, M.P.; McIlwrath, S.L.; Xie, J.; Cheng, C.; Qiao, J.; Tarr, D.E.; Sluka, K.A.; Brennan, T.J.; Lewin, G.R.; Welsh, M.J. The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron 2001, 32, 1071–1083. [Google Scholar] [CrossRef]
- Sluka, K.A.; Price, M.P.; Breese, N.M.; Stucky, C.L.; Wemmie, J.A.; Welsh, M.J. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain 2003, 106, 229–239. [Google Scholar] [CrossRef]
- Andreev, Y.A.; Vassilevski, A.A.; Kozlov, S.A. Molecules to selectively target receptors for treatment of pain and neurogenic inflammation. Recent Pat Inflamm. Allergy Drug Discov. 2012, 6, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, R.; Bassilana, F.; de Weille, J.; Champigny, G.; Heurteaux, C.; Lazdunski, M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J. Biol. Chem. 1997, 272, 20975–20978. [Google Scholar] [CrossRef] [PubMed]
- Voilley, N.; de Weille, J.; Mamet, J.; Lazdunski, M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J. Neurosci. 2001, 21, 8026–8033. [Google Scholar] [CrossRef] [PubMed]
- Leng, T.; Lin, J.; Cottrell, J.E.; Xiong, Z.-G. Subunit and Frequency-Dependent Inhibition of Acid Sensing Ion Channels by Local Anesthetic Tetracaine. Mol. Pain 2013, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Dubé, G.R.; Lehto, S.G.; Breese, N.M.; Baker, S.J.; Wang, X.; Matulenko, M.A.; Honoré, P.; Stewart, A.O.; Moreland, R.B.; Brioni, J.D. Electrophysiological and in vivo characterization of A-317567, a novel blocker of acid sensing ion channels. Pain 2005, 117, 88–96. [Google Scholar] [CrossRef]
- Liu, T.-T.; Qu, Z.-W.; Qiu, C.-Y.; Qiu, F.; Ren, C.; Gan, X.; Peng, F.; Hu, W.-P. Inhibition of acid-sensing ion channels by levo-tetrahydropalmatine in rat dorsal root ganglion neurons. J. Neurosci. Res. 2015, 93, 333–339. [Google Scholar] [CrossRef]
- He, Q.-L.; Chen, Y.; Qin, J.; Mo, S.-L.; Wei, M.; Zhang, J.-J.; Li, M.-N.; Zou, X.-N.; Zhou, S.-F.; Chen, X.-W.; et al. Osthole, a herbal compound, alleviates nucleus pulposus-evoked nociceptive responses through the suppression of overexpression of acid-sensing ion channel 3 (ASIC3) in rat dorsal root ganglion. Med. Sci. Monit. 2012, 18, BR229–BR236. [Google Scholar] [CrossRef] [Green Version]
- Dubinnyi, M.A.; Osmakov, D.I.; Koshelev, S.G.; Kozlov, S.A.; Andreev, Y.A.; Zakaryan, N.A.; Dyachenko, I.A.; Bondarenko, D.A.; Arseniev, A.S.; Grishin, E.V. Lignan from Thyme Possesses Inhibitory Effect on ASIC3 Channel Current. J. Biol. Chem. 2012, 287, 32993–33000. [Google Scholar] [CrossRef] [Green Version]
- Diochot, S.; Baron, A.; Rash, L.D.; Deval, E.; Escoubas, P.; Scarzello, S.; Salinas, M.; Lazdunski, M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. Embo J. 2004, 23, 1516–1525. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, S.A.; Osmakov, D.I.; Andreev, Y.A.; Koshelev, S.G.; Gladkikh, I.N.; Monastyrnaya, M.M.; Kozlovskaya, E.P.; Grishin, E.V. A sea anemone polypeptide toxin inhibiting the ASIC3 acid-sensitive channel. Russ. J. Bioorganic Chem. 2012, 38, 578–583. [Google Scholar] [CrossRef]
- Osmakov, D.I.; Kozlov, S.A.; Andreev, Y.A.; Koshelev, S.G.; Sanamyan, N.P.; Sanamyan, K.E.; Dyachenko, I.A.; Bondarenko, D.A.; Murashev, A.N.; Mineev, K.S.; et al. Sea anemone peptide with uncommon β-hairpin structure inhibits acid-sensing ion channel 3 (ASIC3) and reveals analgesic activity. J. Biol. Chem. 2013, 288, 23116–23127. [Google Scholar] [CrossRef]
- Deval, E.; Noël, J.; Gasull, X.; Delaunay, A.; Alloui, A.; Friend, V.; Eschalier, A.; Lazdunski, M.; Lingueglia, E. Acid-sensing ion channels in postoperative pain. J. Neurosci. 2011, 31, 6059–6066. [Google Scholar] [CrossRef] [PubMed]
- Deval, E.; Noel, J.; Lay, N.; Alloui, A.; Diochot, S.; Friend, V.; Jodar, M.; Lazdunski, M.; Lingueglia, E. ASIC3, a sensor of acidic and primary inflammatory pain. Embo J. 2008, 27, 3047–3055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, J.; Spencer, R.H.; Garsky, V.M.; Liang, A.; Leitl, M.D.; Cato, M.J.; Cook, S.P.; Kane, S.; Urban, M.O. Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br. J. Pharmacol. 2010, 161, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Osmakov, D.I.; Koshelev, S.G.; Andreev, Y.A.; Dyachenko, I.A.; Bondarenko, D.A.; Murashev, A.N.; Grishin, E.V.; Kozlov, S.A. Conversed mutagenesis of an inactive peptide to ASIC3 inhibitor for active sites determination. Toxicon 2016, 116, 11–16. [Google Scholar] [CrossRef]
- Osmakov, D.I.; Koshelev, S.G.; Andreev, Y.A.; Kozlov, S.A. Endogenous isoquinoline alkaloids agonists of acid-sensing ion channel type 3. Front. Mol. Neurosci. 2017, 10, 282. [Google Scholar] [CrossRef]
- Nagakura, Y.; Okada, M.; Kohara, A.; Kiso, T.; Toya, T.; Iwai, A.; Wanibuchi, F.; Yamaguchi, T. Allodynia and Hyperalgesia in Adjuvant-Induced Arthritic Rats: Time Course of Progression and Efficacy of Analgesics. J. Pharmacol. Exp. Ther. 2003, 306, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Parasar, D.; Sagar, A.; Choudhary, V.; Chopra, B.S.; Garg, R.; Ashish Khatri, N. Analgesic and Anti-Inflammatory Properties of Gelsolin in Acetic Acid Induced Writhing, Tail Immersion and Carrageenan Induced Paw Edema in Mice. PLoS ONE 2015, 10, e0135558. [Google Scholar] [CrossRef]
- Blanchard, M.G.; Rash, L.D.; Kellenberger, S. Inhibition of voltage-gated Na(+) currents in sensory neurones by the sea anemone toxin APETx2. Br. J. Pharmacol. 2012, 165, 2167–2177. [Google Scholar] [CrossRef]
- Peigneur, S.; Beress, L.; Moller, C.; Mari, F.; Forssmann, W.-G.; Tytgat, J. A natural point mutation changes both target selectivity and mechanism of action of sea anemone toxins. FASEB J. 2012, 26, 5141–5151. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.E.; Cristofori-Armstrong, B.; Anangi, R.; Rosengren, K.J.; Lau, C.H.Y.; Mobli, M.; Brust, A.; Alewood, P.F.; King, G.F.; Rash, L.D. Understanding the molecular basis of toxin promiscuity: The analgesic sea anemone peptide APETx2 interacts with acid-sensing ion channel 3 and hERG channels via overlapping pharmacophores. J. Med. Chem. 2014, 57, 9195–9203. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.P.; Saez, N.J.; Cristofori-Armstrong, B.; Anangi, R.; King, G.F.; Smith, M.T.; Rash, L.D. Inhibition of acid-sensing ion channels by diminazene and APETx2 evoke partial and highly variable antihyperalgesia in a rat model of inflammatory pain. Br. J. Pharmacol. 2018, 175, 2204–2218. [Google Scholar] [CrossRef] [PubMed]
- Hiasa, M.; Okui, T.; Allette, Y.M.; Ripsch, M.S.; Sun-Wada, G.-H.; Wakabayashi, H.; Roodman, G.D.; White, F.A.; Yoneda, T. Bone Pain Induced by Multiple Myeloma Is Reduced by Targeting V-ATPase and ASIC3. Cancer Res. 2017, 77, 1283–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Sekiguchi, M.; Konno, S. Effect of an Acid-sensing Ion Channels Inhibitor on Pain-related Behavior by Nucleus Pulposus Applied on the Nerve Root in Rats. Spine 2017, 42, E633–E641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Long, H.; Ma, W.; Liao, L.; Yang, X.; Zhou, Y.; Shan, D.; Huang, R.; Jian, F.; Wang, Y.; et al. The role of periodontal ASIC3 in orofacial pain induced by experimental tooth movement in rats. Eur. J. Orthod. 2016, 38, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.X.; Cao, Y.; Ding, T.T.; Fu, K.Y.; Li, Y.; Xie, Q.F. Role of TRPV1 and ASIC3 channels in experimental occlusal interference-induced hyperalgesia in rat masseter muscle. Eur. J. Pain 2016, 20, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Tu, W.; Wen, J.; Zhou, H.; Chen, X.; Zhao, G.; Jiang, Q. The selective ASIC3 inhibitor APETx2 alleviates gastric mucosal lesion in the rat. Pharmazie 2014, 69, 542–546. [Google Scholar]
- Izumi, M.; Ikeuchi, M.; Ji, Q.; Tani, T. Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. J. Biomed. Sci. 2012, 19, 77. [Google Scholar] [CrossRef] [Green Version]
- Yen, L.-T.; Hsieh, C.-L.; Hsu, H.-C.; Lin, Y.-W. Targeting ASIC3 for Relieving Mice Fibromyalgia Pain: Roles of Electroacupuncture, Opioid, and Adenosine. Sci. Rep. 2017, 7, 46663. [Google Scholar] [CrossRef]
- Gregory, N.S.; Brito, R.G.; Fusaro, M.C.G.O.; Sluka, K.A. ASIC3 Is Required for Development of Fatigue-Induced Hyperalgesia. Mol. Neurobiol. 2016, 53, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Blain, R.B. The hormesis database: The occurrence of hormetic dose responses in the toxicological literature. Regul. Toxicol. Pharmacol. 2011, 61, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Cookman, C.J.; Belcher, S.M. Classical nuclear hormone receptor activity as a mediator of complex concentration response relationships for endocrine active compounds. Curr. Opin. Pharmacol. 2014, 19, 112–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Materazzi, S.; Benemei, S.; Fusi, C.; Gualdani, R.; De Siena, G.; Vastani, N.; Andersson, D.A.; Trevisan, G.; Moncelli, M.R.; Wei, X.; et al. Parthenolide inhibits nociception and neurogenic vasodilatation in the trigeminovascular system by targeting the TRPA1 channel. Pain 2013, 154, 2750–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logashina, Y.A.; Mosharova, I.V.; Korolkova, Y.V.; Shelukhina, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Kozlov, S.A.; Stensvåg, K.; et al. Peptide from Sea Anemone Metridium senile Affects Transient Receptor Potential Ankyrin-repeat 1 (TRPA1) Function and Produces Analgesic Effect. J. Biol. Chem. 2017, 292, 2992–3004. [Google Scholar] [CrossRef] [PubMed]
- Logashina, Y.A.; Solstad, R.G.; Mineev, K.S.; Korolkova, Y.V.; Mosharova, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Arseniev, A.S.; et al. New Disulfide-Stabilized Fold Provides Sea Anemone Peptide to Exhibit Both Antimicrobial and TRPA1 Potentiating Properties. Toxins 2017, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, M.V.; Dorofeeva, N.A.; Komarova, M.S.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Grishin, E.V.; Tikhonov, D.B.; Kozlov, S.A. TRPV1 activation power can switch an action mode for its polypeptide ligands. PLoS ONE 2017, 12, e0177077. [Google Scholar] [CrossRef] [PubMed]
- Andreev, Y.A.; Kozlov, S.A.; Korolkova, Y.V.; Dyachenko, I.A.; Bondarenko, D.A.; Skobtsov, D.I.; Murashev, A.N.; Kotova, P.D.; Rogachevskaja, O.A.; Kabanova, N.V.; et al. Polypeptide modulators of TRPV1 produce analgesia without hyperthermia. Mar. Drugs 2013, 11, 5100–5115. [Google Scholar] [CrossRef] [PubMed]
- Mazzuca, M.; Heurteaux, C.; Alloui, A.; Diochot, S.; Baron, A.; Voilley, N.; Blondeau, N.; Escoubas, P.; Gélot, A.; Cupo, A.; et al. A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat. Neurosci. 2007, 10, 943–945. [Google Scholar] [CrossRef]
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef]
- Sluka, K.A.; Gregory, N.S. The dichotomized role for acid sensing ion channels in musculoskeletal pain and inflammation. Neuropharmacology 2015, 94, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, M.W.; Beeton, C.; Galea, C.A.; Smith, B.J.; Chi, V.; Monaghan, K.P.; Garcia, A.; Rangaraju, S.; Giuffrida, A.; Plank, D.; et al. Engineering a stable and selective peptide blocker of the Kv1.3 channel in T lymphocytes. Mol. Pharmacol. 2009, 75, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhou, Q.T.; Chan, H.K.; Larson, I.C.; Pennington, M.W.; Morales, R.A.V.; Boyd, B.J.; Norton, R.S.; Nicolazzo, J.A. Pulmonary Delivery of the Kv1.3-Blocking Peptide HsTX1[R14A] for the Treatment of Autoimmune Diseases. J. Pharm. Sci. 2016, 105, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.A.; Ernst, C.C.; Kramer, W.G.; Madden, D.; Lang, E.; Liao, E.; Lacouture, P.G.; Ramaiya, A.; Carr, D.B. Pharmacokinetics of Diclofenac and Hydroxypropyl-β-Cyclodextrin (HPβCD) Following Administration of Injectable HPβCD-Diclofenac in Subjects with Mild to Moderate Renal Insufficiency or Mild Hepatic Impairment. Clin. Pharmacol. Drug Dev. 2018, 7, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Deval, E.; Baron, A.; Lingueglia, E.; Mazarguil, H.; Zajac, J.-M.; Lazdunski, M. Effects of neuropeptide SF and related peptides on acid sensing ion channel 3 and sensory neuron excitability. Neuropharmacology 2003, 44, 662–671. [Google Scholar] [CrossRef]
- Yagi, J.; Wenk, H.N.; Naves, L.; McCleskey, E.W. Sustained currents through ASIC3 ion channels at the modest pH changes that occur during myocardial ischemia. Circ. Res. 2006, 99, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Yen, Y.-T.; Tu, P.-H.; Chen, C.-J.; Lin, Y.-W.; Hsieh, S.-T.; Chen, C.-C. Role of acid-sensing ion channel 3 in sub-acute-phase inflammation. Mol. Pain 2009, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Anangi, R.; Rash, L.D.; Mobli, M.; King, G.F. Functional Expression in Escherichia coli of the Disulfide-Rich Sea Anemone Peptide APETx2, a Potent Blocker of Acid-Sensing Ion Channel 3. Mar. Drugs 2012, 10, 1605–1618. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antagonist | Transient Currents | Sustained Currents | ||
|---|---|---|---|---|
| IC50 (µM) | Hill Coefficient | IC50 (µM) | Hill Coefficient | |
| APETx2 | 0.344 ± 0.080 | 1.5 ± 0.2 | - | - |
| Ugr 9-1 | 9.1 ± 0.9 | 1.86 ± 0.16 | 1.88 ± 0.36 | 1.74 ± 0.51 |
| sevanol | 331 ± 21 | 1.98 ± 0.24 | 259 ± 39 | 2.4 ± 0.4 |
| diclofenac | - | - | 856 ± 106 | 1.35 ± 0.21 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreev, Y.A.; Osmakov, D.I.; Koshelev, S.G.; Maleeva, E.E.; Logashina, Y.A.; Palikov, V.A.; Palikova, Y.A.; Dyachenko, I.A.; Kozlov, S.A. Analgesic Activity of Acid-Sensing Ion Channel 3 (ASIС3) Inhibitors: Sea Anemones Peptides Ugr9-1 and APETx2 versus Low Molecular Weight Compounds. Mar. Drugs 2018, 16, 500. https://doi.org/10.3390/md16120500
Andreev YA, Osmakov DI, Koshelev SG, Maleeva EE, Logashina YA, Palikov VA, Palikova YA, Dyachenko IA, Kozlov SA. Analgesic Activity of Acid-Sensing Ion Channel 3 (ASIС3) Inhibitors: Sea Anemones Peptides Ugr9-1 and APETx2 versus Low Molecular Weight Compounds. Marine Drugs. 2018; 16(12):500. https://doi.org/10.3390/md16120500
Chicago/Turabian StyleAndreev, Yaroslav A., Dmitry I. Osmakov, Sergey G. Koshelev, Ekaterina E. Maleeva, Yulia A. Logashina, Victor A. Palikov, Yulia A. Palikova, Igor A. Dyachenko, and Sergey A. Kozlov. 2018. "Analgesic Activity of Acid-Sensing Ion Channel 3 (ASIС3) Inhibitors: Sea Anemones Peptides Ugr9-1 and APETx2 versus Low Molecular Weight Compounds" Marine Drugs 16, no. 12: 500. https://doi.org/10.3390/md16120500
APA StyleAndreev, Y. A., Osmakov, D. I., Koshelev, S. G., Maleeva, E. E., Logashina, Y. A., Palikov, V. A., Palikova, Y. A., Dyachenko, I. A., & Kozlov, S. A. (2018). Analgesic Activity of Acid-Sensing Ion Channel 3 (ASIС3) Inhibitors: Sea Anemones Peptides Ugr9-1 and APETx2 versus Low Molecular Weight Compounds. Marine Drugs, 16(12), 500. https://doi.org/10.3390/md16120500