Transcriptomic Profiling and Gene Disruption Revealed that Two Genes Related to PUFAs/DHA Biosynthesis May be Essential for Cell Growth of Aurantiochytrium sp.



Abstract
:1. Introduction
2. Results and Discussion
2.1. De Novo Assembly and Functional Annotation of Aurantiochytrium Transcriptome
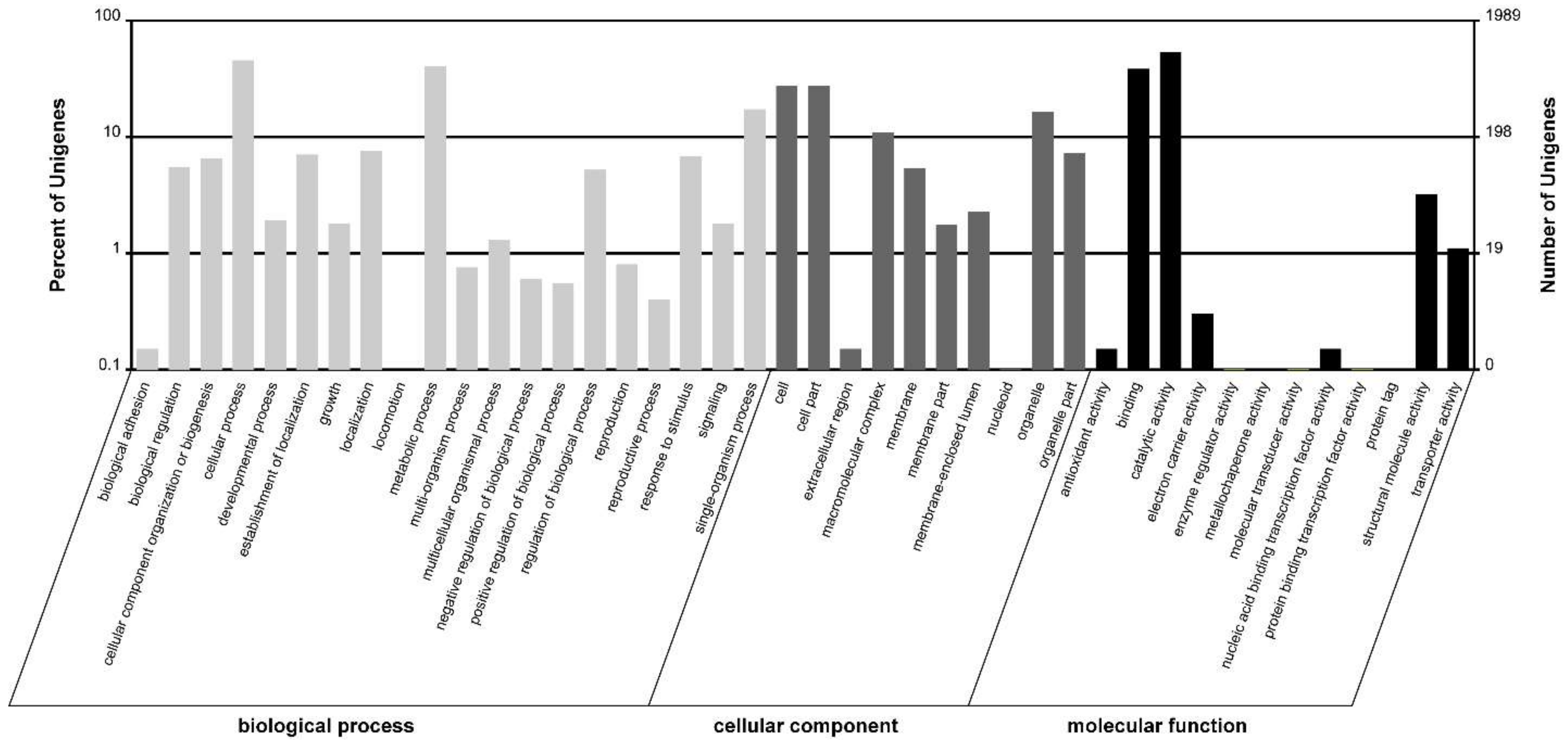
2.2. Function Classification and Pathway Analysis
2.3. Putative Genes Involved in DHA Biosynthesis
2.4. The Relative Expression Level of Putative Key Genes Involved in FAS or PKS Pathways under Various Cultivation Conditions
2.5. Characterization of ΔPH or ΔDH Aurantiochytrium sp. Mutants
3. Materials and Methods
3.1. Microbial Material
3.2. RNA Extraction, cDNA Construction, and RNA-seq
3.3. Transcriptome Assembly and Analysis
3.4. Candidate Genes
3.5. Quantitative Real-Time PCR (qRT-PCR)
3.6. Antibiotics Screening
3.7. Construction of HygR and NeoR Recombination Cassettes
3.8. Transformation of Aurantiochytrium sp. PKU#SW7
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sparrow, F.K. Biological observations on the marine fungi of woods hole waters. Biol. Bull. 1936, 70, 236–263. [Google Scholar] [CrossRef]
- Raghukumar, S.; Balasubramanian, R. Occurrence of thraustochytrids fungi in coral and coral mucus. Indian J. Mar. Sci. 1991, 20, 176–181. [Google Scholar]
- Bongiorni, L. Thraustochytrids, a neglected component of organic matter decomposition and food webs in marine sediments. In Biology of Marine Fungi; Raghukumar, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 53, pp. 1–13. [Google Scholar]
- Raghukumar, S. Bacterivory: A novel dual role for thraustochytrids in the sea. Mar. Biol. 1992, 113, 165–169. [Google Scholar] [CrossRef]
- Bongiorni, L.; Pusceddu, A.; Danovaro, R. Enzymatic activities of epiphytic and benthic thraustochytrids involved in organic matter degradation. Aquat. Microb. Ecol. 2005, 41, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.R.; Nichols, P.D.; Carter, C.G. Replacement of fish oil with thraustochytrid Schizochytrium sp. L. oil in Atlantic salmon parr (Salmo salar L.) diets. Comp. Biochem. Phys. A 2007, 148, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.R.; Lutzu, G.A.; Alam, A.; Sarker, P.; Kabir Chowdhury, M.A.; Parsaeimehr, A.; Liang, Y.; Daroch, M. Microalgae in aquafeeds for a sustainable aquaculture industry. J. Appl. Phycol. 2018, 30, 197–213. [Google Scholar] [CrossRef]
- Lewis, T.E.; Nichols, P.D.; McMeekin, T.A. The biotechnological potential of thraustochytrids. Mar. Biotechnol. 1999, 1, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Raghukumar, S. Thraustochytrid Marine Protists: Production of PUFAs and Other Emerging Technologies. Mar. Biotechnol. 2008, 10, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Wilkens, S.; Adcock, J.L.; Puri, M.; Barrow, C.J. Pollen baiting facilitates the isolation of marine thraustochytrids with potential in omega-3 and biodiesel production. J. Ind. Microbiol. Biot. 2013, 40, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.J.L.; Dunstan, G.A.; Abell, G.C.J.; Clementson, L.A.; Blackburn, S.I.; Nichols, P.D.; Koutoulis, A. Biodiscovery of new Australian thraustochytrids for production of biodiesel and long-chain omega-3 oils. Appl. Microbiol. Biotechnol. 2012, 93, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Marchan, L.F.; Chang, K.J.L.; Nichols, P.D.; Mitchell, W.J.; Polglase, J.L.; Gutierrez, T. Taxonomy, ecology and biotechnological applications of thraustochytrids: A review. Biotechnol. Adv. 2018, 36, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, T.; Tanaka, S.; Yokochi, T.; Nakahara, T.; Higashihara, T. Production of high yields of docosahexaenoic acid by Schizochytrium sp. strain SR21. J. Am. Oil Chem. Soc. 1997, 74, 1431–1434. [Google Scholar] [CrossRef]
- Chi, Z.; Liu, Y.; Frear, C.; Chen, S. Study of a two-stage growth of DHA-producing marine algae Schizochytrium limacinum SR21 with shifting dissolved oxygen level. Appl. Microbiol. Biotechnol. 2009, 81, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Chaung, K.-C.; Chu, C.-Y.; Su, Y.-M.; Chen, Y.-M. Effect of culture conditions on growth, lipid content, and fatty acid composition of Aurantiochytrium mangrovei strain BL10. AMB Express 2012, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, J.; Li, J.; Daroch, M.; Cheng, J.J. Efficient production of triacylglycerols rich in docosahexaenoic acid (DHA) by osmo-heterotrophic marine protists. Appl. Microbiol. Biotechnol. 2014, 98, 9643–9652. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Sakaguchi, K.; Hamaguchi, R.; Kobayashi, T.; Abe, E.; Hama, Y.; Hayashi, M.; Honda, D.; Okita, Y.; Sugimoto, S. Analysis of Δ12-fatty acid desaturase function revealed that two distinct pathways are active for the synthesis of PUFAs in T. aureum ATCC 34304. J. Lipid Res. 2012, 53, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Lippmeier, J.C.; Crawford, K.S.; Owen, C.B.; Rivas, A.A.; Metz, J.G.; Apt, K.E. Characterization of Both Polyunsaturated Fatty Acid Biosynthetic Pathways in Schizochytrium sp. Lipids 2009, 44, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Nagano, N.; Sakaguchi, K.; Taoka, Y.; Okita, Y.; Honda, D.; Ito, M.; Hayashi, M. Detection of Genes Involved in Fatty Acid Elongation and Desaturation in Thraustochytrid Marine Eukaryotes. J. Oleo Sci. 2011, 60, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu Xiao, H.H.; MacKenzie Samuel, L. Identification of a Δ4 fatty acid desaturase from thraustochytrium sp. involved in the biosynthesis of docosahexanoic acid by heterologous expression in saccharomyces cerevisiae and brassica juncea. J. Biol. Chem. 2001, 276, 31561–31566. [Google Scholar]
- Metz, J.G.; Roessler, P.; Facciotti, D.; Levering, C.; Dittrich, F.; Lassner, M.; Valentine, R.; Lardizabal, K.; Domergue, F.; Yamada, A. Production of polyunsaturated fatty acids by polyketide synthases in both prokaryotes and eukaryotes. Science 2001, 293, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Hertwech, C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. 2009, 48, 4688–4716. [Google Scholar] [CrossRef] [PubMed]
- Ratledge, C. Fatty acid biosynthesis in microorganisms being used for Single Cell Oil production. Biochimie 2004, 86, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Singh, P.; Sun, Y.; Luan, S.; Wang, G. Culturable diversity and biochemical features of thraustochytrids from coastal waters of Southern China. Appl. Microbiol. Biotechnol. 2014, 98, 3241–3255. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Tan, Y.; Cui, G.; Feng, Y.; Cui, Q.; Song, X. Transcriptome and gene expression analysis of DHA producer Aurantiochytrium under low temperature conditions. Sci. Rep. 2015, 5, 14446. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhou, P.-P.; Zhang, M.; Zhu, Y.-M.; Wang, X.-P.; Luo, X.-A.; Bao, Z.-D.; Yu, L.-J. Transcriptome analysis reveals that up-regulation of the fatty acid synthase gene promotes the accumulation of docosahexaenoic acid in Schizochytrium sp. S056 when glycerol is used. Algal Res. 2016, 15, 83–92. [Google Scholar] [CrossRef]
- Li, J.; Han, D.; Wang, D.; Ning, K.; Jia, J.; Wei, L.; Jing, X.; Huang, S.; Chen, J.; Li, Y.; Hu, Q.; Xu, J. Choreography of transcriptomes and lipidomes of Nannochloropsis reveals the mechanisms of oil synthesis in microalgae. Plant Cell 2014, 26, 1645–1665. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Deng, Z.; Qin, B.; Liu, X.; Men, Z. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genom. 2012, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Qi, X.; Wang, L.; Zhang, Y.; Hua, W.; Li, D.; Lv, H.; Zhang, X. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom. 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly, annotation, and marker discovery. BMC Genom. 2010, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jiang, X.; Zhang, X.; Chen, W.; Tian, B.; Shu, Z.; Hu, S. Expressed sequence tag analysis of marine fungus Schizochytrium producing docosahexaenoic acid. J. Biotechnol. 2008, 138, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Barrow, C.J.; Puri, M. Omega-3 biotechnology: Thraustochytrids as a novel source of omega-3 oils. Biotechnol. Adv. 2012, 30, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Ryu, B.-G.; Kim, K.; Kim, J.; Han, J.-I.; Yang, J.-W. Use of organic waste from the brewery industry for high-density cultivation of the docosahexaenoic acid-rich microalga, Aurantiochytrium sp. KRS101. Bioresour. Technol. 2013, 129, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Raghukumar, S.; Sambaiah, K.; Kumon, Y.; Nakahara, T. Docosahexaenoic acid accumulation in thraustochytrids: Search for the rationale. Mar. Biol. 2007, 151, 1657–1664. [Google Scholar] [CrossRef]
- Gladyshev, M.I.; Sushchik, N.N.; Makhutova, O.N. Production of EPA and DHA in aquatic ecosystems and their transfer to the land. Prostag. Other Lipid Mediat. 2013, 107, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Damare, V.; Raghukumar, S. Morphology and physiology of the marine straminipilan fungi, the aplanochytrids isolated from the equatorial Indian Ocean. Indian J. Mar. Sci. 2006, 35, 326–340. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; Yamanishi, Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 99, 138–148. [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.M.; Guarente, L. High-efficiency transformation of yeast by electroporation. Method Enzymol. 1991, 194, 182–187. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Putative Enzymes | Number of Unigenes |
|---|---|
| Ketoacyl synthase | 8 |
| 3-oxoacyl-[acyl carrier protein] synthase | 3 |
| Keto reductase | 15 |
| Malonyl CoA-acyl carrier protein transacylase | 1 |
| Malonyl transferase | 3 |
| Dehydrase/isomerase | 2 |
| Enoyl reductase | 4 |
| Hydroxyacyl dehydrogenase | 6 |
| 3-hydroxydecanoyl-[acyl carrier protein] dehydratases | 4 |
| Phosphopantetheinyl transferase | 2 |
| Primer Name | Sequence 5′→3′ | Amplicon Length |
|---|---|---|
| PH-LA-for | CTATAGGGAATATTAAGCTCGCCCTTCTTAGTCTCCGCTTGCGTCGTC | 575 bp |
| PH-LA-rev | GTAGCAGTGAGTTCGGGCTTCTTCATCGCCAGGTTGTAGGCAATGAGG | |
| PH-RA-for | CTACTCGCCCTCGCGCTAAGGAGTAAGGTTTGTCATTACGCCAGTTGT | 584 bp |
| PH-RA-rev | AATGGGTGACCTCGAAGCTCGCCCTCTACGCCACCAGTCCTAACAAG | |
| DH-LA-for | CTATAGGGAATATTAAGCTCGCCCTTTTCTAAGGCCGGCTATGTATGC | 559 bp |
| DH-LA-rev | ACCGTCCTGCTCAATAGCAGACATGTCCAGGAAGCATCCGACACAC | |
| DH-RA-for | CCGTCTCCTCGATGAGTTTTTTTAACTTTAAGAAAGGAAGCAATGAGCC | 545 bp |
| DH-RA-rev | AATGGGTGACCTCGAAGCTCGCCCTCTAAATGATACAGCCTTTGTTCGT | |
| PHck-for | CTTCTTAGTCTCCGCTTGCGTCGTC | ΔPH-SW7: 2237 bp SW7: 3286 bp |
| PHck-rev | CTCTACGCCACCAGTCCTAACAAG | |
| DHck-for | TTCTAAGGCCGGCTATGTATGC | ΔDH-SW7: 1902 bp SW7: 2484 bp |
| DHck-rev | TCTAAATGATACAGCCTTTGTTCG | |
| Hygro-for | ATGAAGAAGCCCGAACTCACTGC | 1078 bp |
| Hygro-rev | TTACTCCTTAGCGCGAGGGCGAGTA | |
| NeoR-for | ATCTCATGACCAAAATCCCTTAACGTG | 798 bp |
| NeoR-rev | TTAAAAAAACTCATCGAGGAGACGGT | |
| BF | ATGGCTGACGACGAAGTTCAAGC | 175 bp |
| BR | CCTCATCACCGACATAGGC | |
| PHF | TGGTGCTAGGAGCAACGTTGCTAG | 139 bp |
| PHR | TTCTGGCCTGAAGCTCAACAACTC | |
| DHF | GGCAATCATAATAAGCTTCCTTTCACCTTGC | 133 bp |
| DHR | CATCTTTAAGAAAGGAAGCAATGAGCCG |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Liu, Y.; Tang, J.; Ma, J.; Cheng, J.J.; Daroch, M. Transcriptomic Profiling and Gene Disruption Revealed that Two Genes Related to PUFAs/DHA Biosynthesis May be Essential for Cell Growth of Aurantiochytrium sp. Mar. Drugs 2018, 16, 310. https://doi.org/10.3390/md16090310
Liang Y, Liu Y, Tang J, Ma J, Cheng JJ, Daroch M. Transcriptomic Profiling and Gene Disruption Revealed that Two Genes Related to PUFAs/DHA Biosynthesis May be Essential for Cell Growth of Aurantiochytrium sp. Marine Drugs. 2018; 16(9):310. https://doi.org/10.3390/md16090310
Chicago/Turabian StyleLiang, Yuanmei, Ying Liu, Jie Tang, Jiong Ma, Jay J. Cheng, and Maurycy Daroch. 2018. "Transcriptomic Profiling and Gene Disruption Revealed that Two Genes Related to PUFAs/DHA Biosynthesis May be Essential for Cell Growth of Aurantiochytrium sp." Marine Drugs 16, no. 9: 310. https://doi.org/10.3390/md16090310
APA StyleLiang, Y., Liu, Y., Tang, J., Ma, J., Cheng, J. J., & Daroch, M. (2018). Transcriptomic Profiling and Gene Disruption Revealed that Two Genes Related to PUFAs/DHA Biosynthesis May be Essential for Cell Growth of Aurantiochytrium sp. Marine Drugs, 16(9), 310. https://doi.org/10.3390/md16090310