Extensive Tandem Duplication Events Drive the Expansion of the C1q-Domain-Containing Gene Family in Bivalves




Abstract
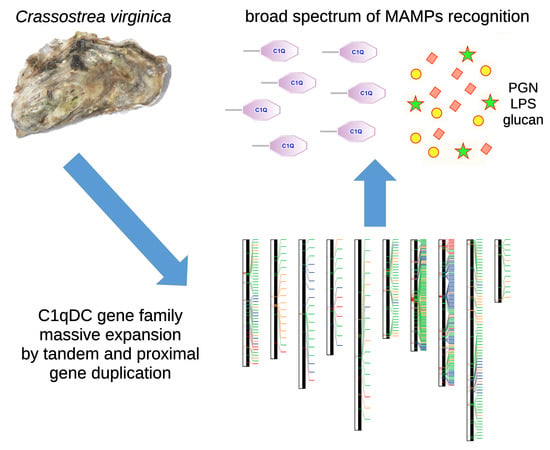
:
1. Introduction
2. Results
2.1. The Repertoire of the Eastern Oyster C1qDC Genes
2.2. Chromosomal Distribution of Oyster C1qDC Genes
2.3. Most Oyster C1qDC Genes Are Tandemly Duplicated
2.4. The Gene Duplication Process Is Still Ongoing and Is Paired with Diversifying Selection
3. Discussion
4. Materials and Methods
4.1. Identification of C1qDC Genes
4.2. Sequence Analysis of C1qDC Genes
4.3. Positive Selection Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gorbushin, A.M. Immune repertoire in the transcriptome of Littorina littorea reveals new trends in lophotrochozoan proto-complement evolution. Dev. Comp. Immunol. 2018, 84, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Pednekar, L.; Reid, K.B.M.; Kishore, U. Complement and non-complement activating functions of C1q: A prototypical innate immune molecule. Innate Immun. 2012, 18, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Tom Tang, Y.; Hu, T.; Arterburn, M.; Boyle, B.; Bright, J.M.; Palencia, S.; Emtage, P.C.; Funk, W.D. The complete complement of C1q-domain-containing proteins in Homo sapiens. Genomics 2005, 86, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Carland, T.M.; Gerwick, L. The C1q domain containing proteins: Where do they come from and what do they do? Dev. Comp. Immunol. 2010, 34, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Guo, X.; Litman, G.W.; Dishaw, L.J.; Zhang, G. Massive expansion and functional divergence of innate immune genes in a protostome. Sci. Rep. 2015, 5, 8693. [Google Scholar] [CrossRef]
- Gerdol, M.; Luo, Y.-J.; Satoh, N.; Pallavicini, A. Genetic and molecular basis of the immune system in the brachiopod Lingula anatina. Dev. Comp. Immunol. 2018, 82, 7–30. [Google Scholar] [CrossRef]
- Gerdol, M.; Manfrin, C.; De Moro, G.; Figueras, A.; Novoa, B.; Venier, P.; Pallavicini, A. The C1q domain containing proteins of the Mediterranean mussel Mytilus galloprovincialis: A widespread and diverse family of immune-related molecules. Dev. Comp. Immunol. 2011, 35, 635–643. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Zhu, Y.; Zhang, G.; Guo, X. Transcriptome analysis reveals a rich gene set related to innate immunity in the Eastern oyster (Crassostrea virginica). Mar. Biotechnol. 2014, 16, 17–33. [Google Scholar] [CrossRef]
- Takeuchi, T.; Koyanagi, R.; Gyoja, F.; Kanda, M.; Hisata, K.; Fujie, M.; Goto, H.; Yamasaki, S.; Nagai, K.; Morino, Y.; et al. Bivalve-specific gene expansion in the pearl oyster genome: Implications of adaptation to a sessile lifestyle. Zool. Lett. 2016, 2, 3. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Y.; Xu, T.; Zhang, Y.; Mu, H.; Zhang, Y.; Lan, Y.; Fields, C.J.; Hui, J.H.L.; Zhang, W.; et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 2017, 1, 0121. [Google Scholar] [CrossRef]
- Powell, D.; Subramanian, S.; Suwansa-Ard, S.; Zhao, M.; O’Connor, W.; Raftos, D.; Elizur, A. The genome of the oyster Saccostrea offers insight into the environmental resilience of bivalves. DNA Res. Int. J. Rapid Publ. Rep. Genes. Genomes 2018, 25, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Mun, S.; Kim, Y.-J.; Markkandan, K.; Shin, W.; Oh, S.; Woo, J.; Yoo, J.; An, H.; Han, K. The Whole-Genome and Transcriptome of the Manila Clam (Ruditapes philippinarum). Genome Biol. Evol. 2017, 9, 1487–1498. [Google Scholar] [CrossRef]
- Gerdol, M.; Venier, P.; Pallavicini, A. The genome of the Pacific oyster Crassostrea gigas brings new insights on the massive expansion of the C1q gene family in Bivalvia. Dev. Comp. Immunol. 2015, 49, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Liu, Z.; Wu, Z.; Han, Z.; Wang, L.; Song, L. A novel globular C1q domain containing protein (C1qDC-7) from Crassostrea gigas acts as pattern recognition receptor with broad recognition spectrum. Fish. Shellfish. Immunol. 2019, 84, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wei, Z.; Shen, Y.; Li, C.; Shao, Y.; Zhang, W.; Zhao, X. A novel C1q-domain-containing protein from razor clam Sinonovacula constricta mediates G-bacterial agglutination as a pattern recognition receptor. Dev. Comp. Immunol. 2018, 79, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, H.; Wang, M.; Zhou, Z.; Wang, W.; Liu, R.; Huang, M.; Yang, C.; Qiu, L.; Song, L. The transcriptomic expression of pattern recognition receptors: Insight into molecular recognition of various invading pathogens in Oyster Crassostrea gigas. Dev. Comp. Immunol. 2019, 91, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Kong, P.; Yang, J.; Zhang, H.; Wang, M.; Zhou, Z.; Qiu, L.; Song, L. A novel C1qDC protein acting as pattern recognition receptor in scallop Argopecten irradians. Fish. Shellfish. Immunol. 2012, 33, 427–435. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Zhang, D.; Jiang, Q.; Sun, R.; Wang, H.; Zhang, H.; Song, L. A novel multi-domain C1qDC protein from Zhikong scallop Chlamys farreri provides new insights into the function of invertebrate C1qDC proteins. Dev. Comp. Immunol. 2015, 52, 202–214. [Google Scholar] [CrossRef]
- Gaboriaud, C.; Juanhuix, J.; Gruez, A.; Lacroix, M.; Darnault, C.; Pignol, D.; Verger, D.; Fontecilla-Camps, J.C.; Arlaud, G.J. The Crystal Structure of the Globular Head of Complement Protein C1q Provides a Basis for Its Versatile Recognition Properties. J. Biol. Chem. 2003, 278, 46974–46982. [Google Scholar] [CrossRef] [Green Version]
- Reid, K.B.M. Complement Component C1q: Historical Perspective of a Functionally Versatile, and Structurally Unusual, Serum Protein. Front. Immunol. 2018, 9, 764. [Google Scholar] [CrossRef]
- Yin, Y.; Huang, J.; Paine, M.L.; Reinhold, V.N.; Chasteen, N.D. Structural characterization of the major extrapallial fluid protein of the mollusc Mytilus edulis: Implications for function. Biochemistry 2005, 44, 10720–10731. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ruan, Z.; Yang, X.; Li, M.; Yang, D. Immune recognition, antimicrobial and opsonic activities mediated by a sialic acid binding lectin from Ruditapes philippinarum. Fish. Shellfish. Immunol. 2019, 93, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, H.; Zhang, D.; Zhang, H.; Wang, L.; Sun, J.; Song, L. A C1q domain containing protein from Crassostrea gigas serves as pattern recognition receptor and opsonin with high binding affinity to LPS. Fish. Shellfish. Immunol. 2015, 45, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kong, N.; Sun, J.; Wang, W.; Li, M.; Gong, C.; Dong, M.; Wang, M.; Wang, L.; Song, L. A C1qDC (CgC1qDC-6) with a collagen-like domain mediates hemocyte phagocytosis and migration in oysters. Dev. Comp. Immunol. 2019, 98, 157–165. [Google Scholar] [CrossRef]
- Gómez-Chiarri, M.; Warren, W.C.; Guo, X.; Proestou, D. Developing tools for the study of molluscan immunity: The sequencing of the genome of the eastern oyster, Crassostrea virginica. Fish. Shellfish. Immunol. 2015, 46, 2–4. [Google Scholar] [CrossRef]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, T.; Ichise, A.; Unno, H.; Goda, S.; Oda, T.; Tateno, H.; Hirabayashi, J.; Sakai, H.; Nakagawa, H. Carbohydrate recognition by the rhamnose-binding lectin SUL-I with a novel three-domain structure isolated from the venom of globiferous pedicellariae of the flower sea urchin Toxopneustes pileolus. Protein Sci. Publ. Protein Soc. 2017, 26, 1574–1583. [Google Scholar] [CrossRef]
- Ozeki, Y.; Matsui, T.; Suzuki, M.; Titani, K. Amino acid sequence and molecular characterization of a D-galactoside-specific lectin purified from sea urchin (Anthocidaris crassispina) eggs. Biochemistry 1991, 30, 2391–2394. [Google Scholar] [CrossRef]
- Carneiro, R.F.; Teixeira, C.S.; de Melo, A.A.; de Almeida, A.S.; Cavada, B.S.; de Sousa, O.V.; da Rocha, B.A.M.; Nagano, C.S.; Sampaio, A.H. L-Rhamnose-binding lectin from eggs of the Echinometra lucunter: Amino acid sequence and molecular modeling. Int. J. Biol. Macromol. 2015, 78, 180–188. [Google Scholar] [CrossRef]
- Freeling, M. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Maere, S.; De Bodt, S.; Raes, J.; Casneuf, T.; Van Montagu, M.; Kuiper, M.; Van de Peer, Y. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. USA. 2005, 102, 5454–5459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusack, B.P.; Wolfe, K.H. Not born equal: Increased rate asymmetry in relocated and retrotransposed rodent gene duplicates. Mol. Biol. Evol. 2007, 24, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Kaessmann, H.; Vinckenbosch, N.; Long, M. RNA-based gene duplication: Mechanistic and evolutionary insights. Nat. Rev. Genet. 2009, 10, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zuo, T.; Peterson, T. Generation of tandem direct duplications by reversed-ends transposition of maize ac elements. PLoS Genet. 2013, 9, e1003691. [Google Scholar] [CrossRef]
- Wagner, A. The fate of duplicated genes: Loss or new function? BioEssays News Rev. Mol. Cell. Dev. Biol. 1998, 20, 785–788. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar] [CrossRef]
- Hahn, M.W. Distinguishing among evolutionary models for the maintenance of gene duplicates. J. Hered. 2009, 100, 605–617. [Google Scholar] [CrossRef]
- Rastogi, S.; Liberles, D.A. Subfunctionalization of duplicated genes as a transition state to neofunctionalization. BMC Evol. Biol. 2005, 5, 28. [Google Scholar] [CrossRef]
- Gaboriaud, C.; Frachet, P.; Thielens, N.M.; Arlaud, G.J. The Human C1q Globular Domain: Structure and Recognition of Non-Immune Self Ligands. Front. Immunol. 2012, 2, 92. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Shrive, A.K.; Greenhough, T.J.; Volanakis, J.E. Topology and structure of the C1q-binding site on C-reactive protein. J. Immunol. Baltim. MD 1950 2001, 166, 3998–4004. [Google Scholar] [CrossRef] [PubMed]
- Gorbushin, A.M. Derivatives of the lectin complement pathway in Lophotrochozoa. Dev. Comp. Immunol. 2019, 94, 35–58. [Google Scholar] [CrossRef] [PubMed]
- McDowell, I.C.; Modak, T.H.; Lane, C.E.; Gomez-Chiarri, M. Multi-species protein similarity clustering reveals novel expanded immune gene families in the eastern oyster Crassostrea virginica. Fish. Shellfish. Immunol. 2016, 53, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdol, M.; Moreira, R.; Cruz, F.; Gómez-Garrido, J.; Vlasova, A.; Rosani, U.; Venier, P.; Naranjo-Ortiz, M.A.; Murgarella, M.; Balseiro, P.; et al. Massive gene presence/absence variation in the mussel genome as an adaptive strategy: First evidence of a pan-genome in Metazoa. BioRxiv 2019, 781377. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Lupas, A.; Van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. BioRxiv 2019, 612903. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A General Empirical Model of Protein Evolution Derived from Multiple Protein Families Using a Maximum-Likelihood Approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinf. Oxf. Engl. 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Armougom, F.; Moretti, S.; Poirot, O.; Audic, S.; Dumas, P.; Schaeli, B.; Keduas, V.; Notredame, C. Expresso: Automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006, 34, W604–W608. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| chr1 | chr2 | chr3 | chr4 | chr5 | chr6 | chr7 | chr8 | chr9 | chr10 | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome size (Mb) | 65.67 | 61.72 | 77.06 | 59.69 | 98.70 | 51.26 | 57.83 | 75.94 | 104.16 | 32.65 | 684.68 |
| Number of C1qDC genes | 45 | 17 | 18 | 12 | 17 | 36 | 123 | 123 | 73 | 12 | 476 |
| C1qDC density (genes/Mb) | 0.67 | 0.28 | 0.23 | 0.28 | 0.17 | 0.70 | 2.13 | 1.63 | 0.70 | 0.37 | 0.70 |
| sghC1q genes | 8 | 0 | 4 | 3 | 1 | 2 | 18 | 63 | 12 | 0 | 111 |
| sC1q-like type II genes | 27 | 6 | 10 | 7 | 12 | 26 | 81 | 36 | 48 | 9 | 262 |
| smultiC1q genes | 3 | 1 | 0 | 2 | 1 | 0 | 1 | 14 | 0 | 0 | 21 |
| sSUEL/C1q | 0 | 10 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 16 |
| Other C1qDc genes | 7 | 0 | 4 | 0 | 3 | 8 | 17 | 10 | 13 | 3 | 63 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerdol, M.; Greco, S.; Pallavicini, A. Extensive Tandem Duplication Events Drive the Expansion of the C1q-Domain-Containing Gene Family in Bivalves. Mar. Drugs 2019, 17, 583. https://doi.org/10.3390/md17100583
Gerdol M, Greco S, Pallavicini A. Extensive Tandem Duplication Events Drive the Expansion of the C1q-Domain-Containing Gene Family in Bivalves. Marine Drugs. 2019; 17(10):583. https://doi.org/10.3390/md17100583
Chicago/Turabian StyleGerdol, Marco, Samuele Greco, and Alberto Pallavicini. 2019. "Extensive Tandem Duplication Events Drive the Expansion of the C1q-Domain-Containing Gene Family in Bivalves" Marine Drugs 17, no. 10: 583. https://doi.org/10.3390/md17100583
APA StyleGerdol, M., Greco, S., & Pallavicini, A. (2019). Extensive Tandem Duplication Events Drive the Expansion of the C1q-Domain-Containing Gene Family in Bivalves. Marine Drugs, 17(10), 583. https://doi.org/10.3390/md17100583