Novel Microcystins from Planktothrix prolifica NIVA-CYA 544 Identified by LC-MS/MS, Functional Group Derivatization and 15N-labeling



Abstract
:1. Introduction
2. Results and Discussion
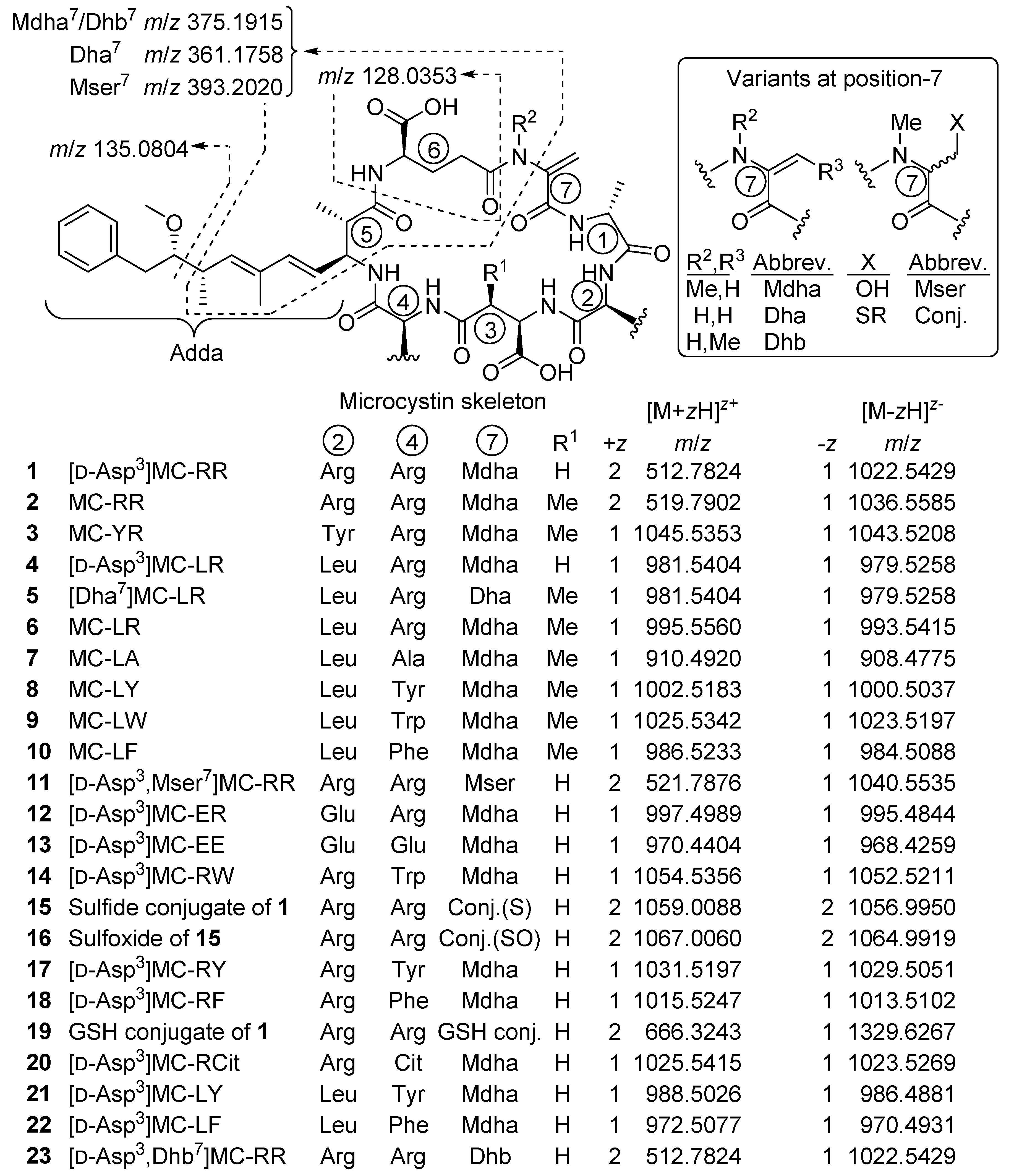
2.1. MCs Profiling of P. prolifica NIVA-CYA 544
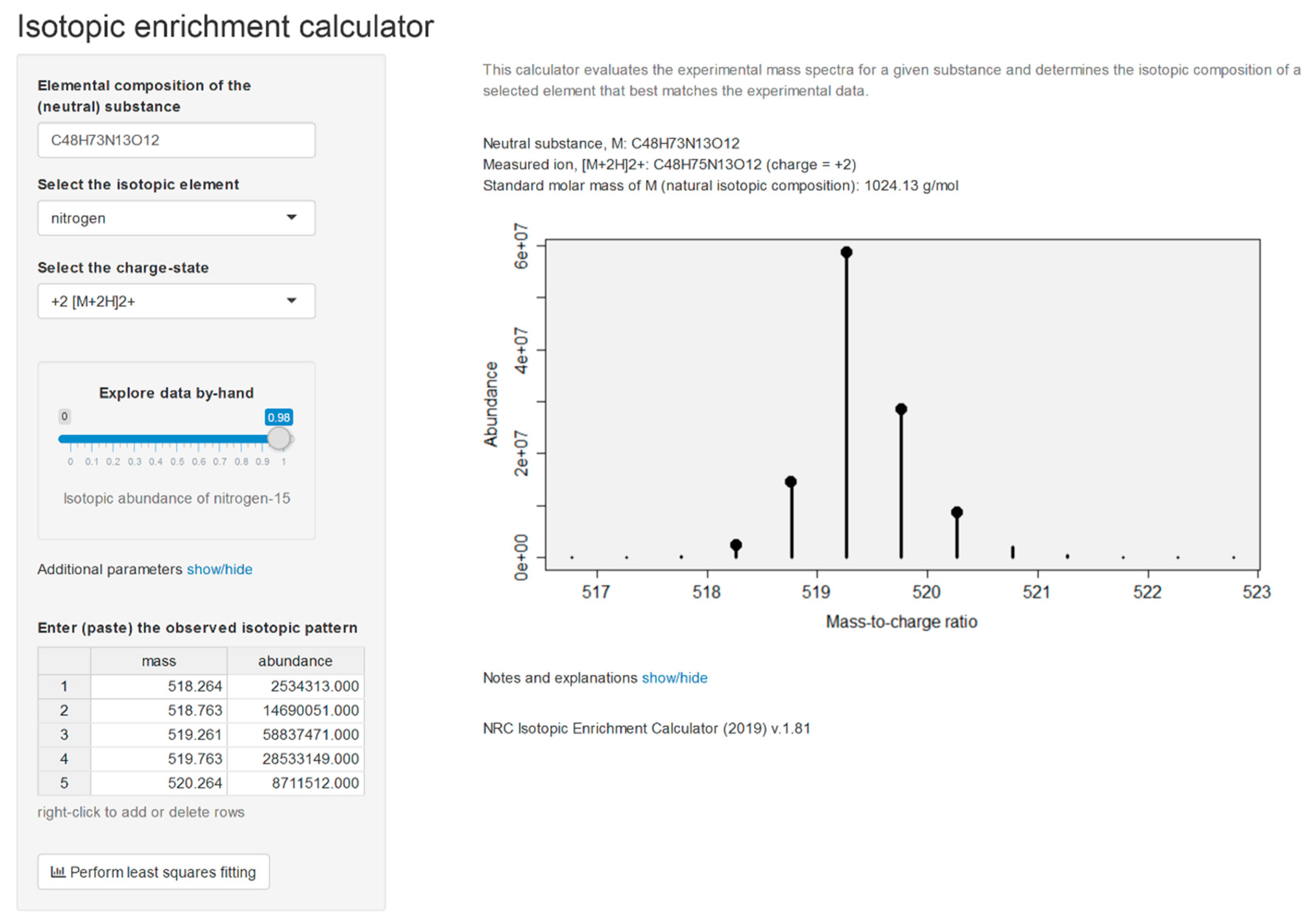
2.2. Isotopic Enrichment Calculations
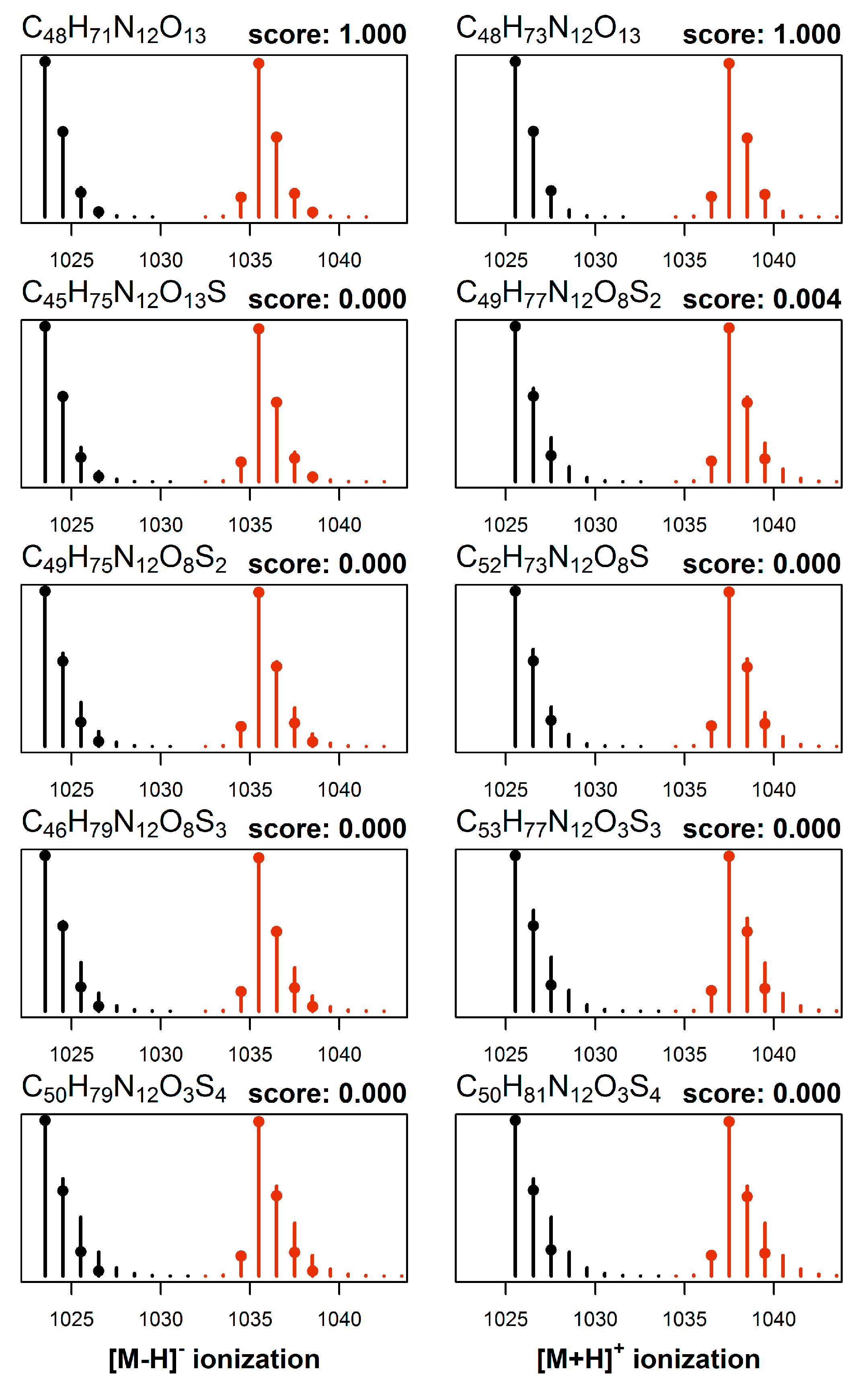
2.3. Elemental Composition Elucidation
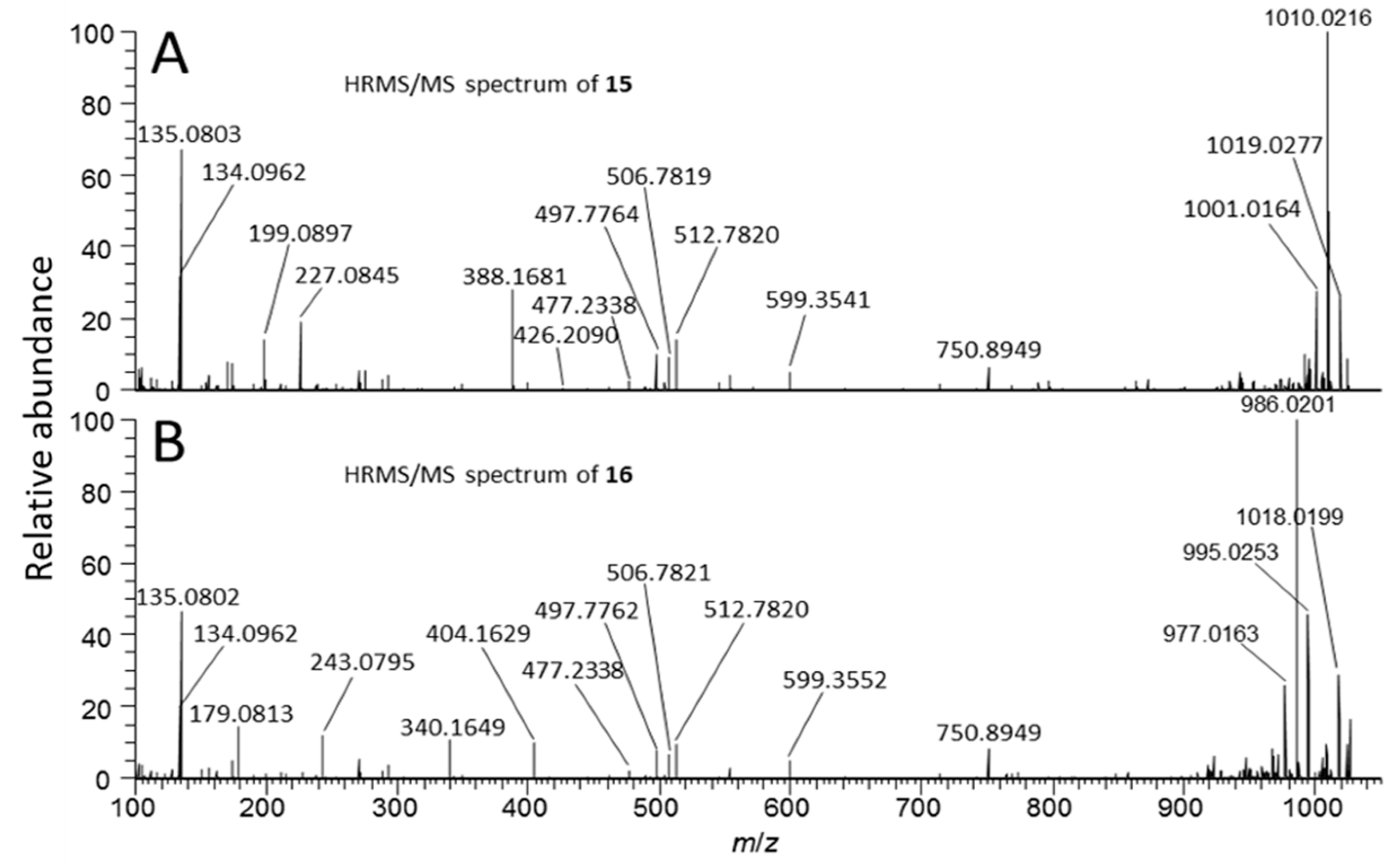
2.4. Identification of MC Congeners
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Cultivation of P. prolifica NIVA-CYA 544 and Extraction of MCs
3.3. Cultivation of P. prolifica NIVA-CYA 544 for 15N-labeling of MCs
3.4. Liquid Chromatography–Mass Spectrometry
3.4.1. LC–HRMS and LC–HRMS/MS (Method A)
3.4.2. LC–HRMS and LC–HRMS/MS (Method B)
3.4.3. LC–ITMS/MS (Method C)
3.5. 2-Mercaptoethanol Derivatization for Mdha7/Dhb7 Differentiation
3.6. Methylation of Carboxylic Acids
3.7. 15N-Incorporation and Molecular Formula Calculations
3.8. Reaction of 1 with Glutathione to Produce 19.
3.9. Oxidation with Sodium Periodate
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Benke, P.I.; Vinay Kumar, M.C.; Pan, D.; Swarup, S. A mass spectrometry-based unique fragment approach for the identification of microcystins. Analyst 2015, 140, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, R.; Li, J. Current research scenario for microcystins biodegradation—A review on fundamental knowledge, application prospects and challenges. Sci. Total Environ. 2017, 595, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W.; Hall, N.S.; Calandrino, E.S. Controlling harmful cyanobacterial blooms in a world experiencing anthropogenic and climatic-induced change. Sci. Total Environ. 2011, 409, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Kudela, R.M.; Mekebri, A.; Crane, D.; Oates, S.C.; Tinker, M.T.; Staedler, M.; Miller, W.A.; Toy-Choutka, S.; Dominik, C.; et al. Evidence for a novel marine harmful algal bloom: Cyanotoxin (microcystin) transfer from land to sea otters. PLoS ONE 2010, 5, e12576. [Google Scholar] [CrossRef]
- Fontanillo, M.; Kohn, M. Microcystins: Synthesis and structure-activity relationship studies toward PP1 and PP2A. Bioorg. Med. Chem. 2018, 26, 1118–1126. [Google Scholar] [CrossRef]
- Carmichael, W.W.; Azevedo, S.M.; An, J.S.; Molica, R.J.; Jochimsen, E.M.; Lau, S.; Rinehart, K.L.; Shaw, G.R.; Eaglesham, G.K. Human fatalities from cyanobacteria: Chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001, 109, 663–668. [Google Scholar] [CrossRef]
- Jochimsen, E.M.; Carmichael, W.W.; An, J.S.; Cardo, D.M.; Cookson, S.T.; Holmes, C.E.; Antunes, M.B.; de Melo Filho, D.A.; Lyra, T.M.; Barreto, V.S.; et al. Liver failure and death after exposure to microcystins at a hemodialysis center in Brazil. N. Engl. J. Med. 1998, 338, 873–878. [Google Scholar] [CrossRef]
- MacKintosh, C.; Beattie, K.A.; Klumpp, S.; Cohen, P.; Codd, G.A. Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett. 1990, 264, 187–192. [Google Scholar] [CrossRef]
- Yoshizawa, S.; Matsushima, R.; Watanabe, M.F.; Harada, K.; Ichihara, A.; Carmichael, W.W.; Fujiki, H. Inhibition of protein phosphatases by microcystins and nodularin associated with hepatotoxicity. J. Cancer Res. Clin. Oncol. 1990, 116, 609–614. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.; Zhang, X.; Xie, P. A review of reproductive toxicity of microcystins. J. Hazard. Mater. 2016, 301, 381–399. [Google Scholar] [CrossRef]
- Chen, L.; Li, S.C.; Guo, X.C.; Xie, P.; Chen, J. The role of GSH in microcystin-induced apoptosis in rat liver: Involvement of oxidative stress and NF-κB. Environ. Toxicol. 2016, 31, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Xie, P.; Li, H.; Hao, L.; Li, G.; Qiu, T.; Liu, Y. Acute effects of microcystins exposure on the transcription of antioxidant enzyme genes in three organs (liver, kidney, and testis) of male Wistar rats. J. Biochem. Mol. Toxicol. 2010, 24, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, G.; Fastner, J.; Erhard, M.; Borner, T.; Dittmann, E. Microcystin biosynthesis in Planktothrix: Genes, evolution, and manipulation. J. Bacteriol. 2003, 185, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.; Santiago-Vazquez, L.; Paul, V. Toxin release in response to oxidative stress and programmed cell death in the cyanobacterium Microcystis aeruginosa. Aquat. Toxicol. 2006, 78, 66–73. [Google Scholar] [CrossRef]
- Botes, D.P.; Kruger, H.; Viljoen, C.C. Isolation and characterization of four toxins from the blue–green alga, Microcystis aeruginosa. Toxicon 1982, 20, 945–954. [Google Scholar] [CrossRef]
- Huisman, J.; Codd, G.A.; Paerl, H.W.; Ibelings, B.W.; Verspagen, J.M.H.; Visser, P.M. Cyanobacterial blooms. Nat. Rev. Microbiol. 2018, 16, 471–483. [Google Scholar] [CrossRef]
- Kleinteich, J.; Puddick, J.; Wood, S.A.; Hildebrand, F.; Laughinghouse, H.I.; Pearce, D.A.; Dietrich, D.R.; Wilmotte, A. Toxic cyanobacteria in Svalbard: Chemical diversity of microcystins detected using a liquid chromatography mass spectrometry precursor ion screening method. Toxins 2018, 10, 147. [Google Scholar] [CrossRef]
- WHO. Guidelines for Drinking-Water Quality: Fourth Edition Incorporating the First Addendum; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- He, H.; Wu, S.; Wahome, P.G.; Bertin, M.J.; Pedone, A.C.; Beauchesne, K.R.; Moeller, P.D.R.; Carter, G.T. Microcystins containing doubly homologated tyrosine residues from a Microcystis aeruginosa bloom: Structures and cytotoxicity. J. Nat. Prod. 2018, 81, 1368–1375. [Google Scholar] [CrossRef]
- Feurstein, D.; Stemmer, K.; Kleinteich, J.; Speicher, T.; Dietrich, D.R. Microcystin congener- and concentration-dependent induction of murine neuron apoptosis and neurite degeneration. Toxicol. Sci. 2011, 124, 424–431. [Google Scholar] [CrossRef]
- Qi, Y.; Bortoli, S.; Volmer, D.A. Detailed study of cyanobacterial microcystins using high performance tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2014, 25, 1253–1262. [Google Scholar] [CrossRef]
- Schmidt, J.R.; Wilhelm, S.W.; Boyer, G.L. The fate of microcystins in the environment and challenges for monitoring. Toxins 2014, 6, 3354–3387. [Google Scholar] [CrossRef] [PubMed]
- Stotts, R.R.; Namikoshi, M.; Haschek, W.M.; Rinehart, K.L.; Carmichael, W.W.; Dahlem, A.M.; Beasley, V.R. Structural modifications imparting reduced toxicity in microcystins from Microcystis spp. Toxicon 1993, 31, 783–789. [Google Scholar] [CrossRef]
- Bortoli, S.; Volmer, D.A. Characterization and identification of microcystins by mass spectrometry. Eur. J. Mass Spectrom. 2014, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Foss, A.J.; Miles, C.O.; Samdal, I.A.; Løvberg, K.E.; Wilkins, A.L.; Rise, F.; Jaabaek, J.A.H.; McGowan, P.C.; Aubel, M.T. Analysis of free and metabolized microcystins in samples following a bird mortality event. Harmful Algae 2018, 80, 117–129. [Google Scholar] [CrossRef]
- Harada, K.I.; Tsuji, K.; Watanabe, M.F.; Kondo, F. Stability of microcystins from cyanobacteria—III. Effect of pH and temperature. Phycologia 1996, 35, 83–88. [Google Scholar] [CrossRef]
- Janssen, E.M. Cyanobacterial peptides beyond microcystins—A review on co-occurrence, toxicity, and challenges for risk assessment. Water Res. 2019, 151, 488–499. [Google Scholar] [CrossRef]
- Shah, S.A.A.; Akhter, N.; Auckloo, B.N.; Khan, I.; Lu, Y.; Wang, K.; Wu, B.; Guo, Y.W. Structural diversity, biological properties and applications of natural products from cyanobacteria. A review. Mar. Drugs 2017, 15, 354. [Google Scholar] [CrossRef]
- Rogers, E.D.; Henry, T.B.; Twiner, M.J.; Gouffon, J.S.; McPherson, J.T.; Boyer, G.L.; Sayler, G.S.; Wilhelm, S.W. Global gene expression profiling in larval zebrafish exposed to microcystin-LR and microcystis reveals endocrine disrupting effects of cyanobacteria. Environ. Sci. Technol. 2011, 45, 1962–1969. [Google Scholar] [CrossRef]
- Stepankova, T.; Ambrozova, L.; Blaha, L.; Giesy, J.P.; Hilscherova, K. In vitro modulation of intracellular receptor signaling and cytotoxicity induced by extracts of cyanobacteria, complex water blooms and their fractions. Aquat. Toxicol. 2011, 105, 497–507. [Google Scholar] [CrossRef]
- Miles, C.O.; Sandvik, M.; Haande, S.; Nonga, H.; Ballot, A. LC–MS analysis with thiol derivatization to differentiate [Dhb7]- from [Mdha7]-microcystins: Analysis of cyanobacterial blooms, Planktothrix cultures and European crayfish from Lake Steinsfjorden, Norway. Environ. Sci. Technol. 2013, 47, 4080–4087. [Google Scholar] [CrossRef]
- Miles, C.O.; Sandvik, M.; Nonga, H.E.; Rundberget, T.; Wilkins, A.L.; Rise, F.; Ballot, A. Thiol derivatization for LC–MS identification of microcystins in complex matrices. Environ. Sci. Technol. 2012, 46, 8937–8944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, M.; Foss, A.J.; Miles, C.O.; Ozen, M.; Demir, N.; Balci, M.; Beach, D.G. Comprehensive multi-technique approach reveals the high diversity of microcystins in field collections and an associated isolate of Microcystis aeruginosa from a Turkish lake. Toxicon 2019, 167, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.O.; Melanson, J.E.; Ballot, A. Sulfide oxidations for LC–MS analysis of methionine-containing microcystins in Dolichospermum flos-aquae NIVA-CYA 656. Environ. Sci. Technol. 2014, 48, 13307–13315. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Strangman, W.K.; Percy, A.; Wright, J.L.C. The biosynthesis of 15N-labeled microcystins and the comparative MS/MS fragmentation of natural abundance and their 15N-labeled congeners using LC–MS/MS. Toxicon 2018, 144, 91–102. [Google Scholar] [CrossRef] [PubMed]
- MacCoss, M.J.; Wu, C.C.; Matthews, D.E.; Yates, J.R., III. Measurement of the isotope enrichment of stable isotope-labeled proteins using high-resolution mass spectra of peptides. Anal. Chem. 2005, 77, 7646–7653. [Google Scholar] [CrossRef] [PubMed]
- Meija, J. An ode to the atomic weights. Nat. Chem. 2014, 6, 749–750. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, A. Efficient calculation of exact fine structure isotope patterns via the multidimensional Fourier transform. Anal. Chem. 2014, 86, 5316–5322. [Google Scholar] [CrossRef]
- Bocker, S.; Letzel, M.C.; Liptak, Z.; Pervukhin, A. SIRIUS: Decomposing isotope patterns for metabolite identification. Bioinformatics 2009, 25, 218–224. [Google Scholar] [CrossRef]
- Pervukhin, A.; Neumann, S. Decomposition of Isotopic Patterns. R package: Rdisop, 1.42. 2017. Available online: https://www.bioconductor.org/packages/release/bioc/html/Rdisop.html (accessed on 7 October 2019).
- Senior, J.K. Partitions and their representative graphs. Am. J. Math. 1951, 73, 663–689. [Google Scholar] [CrossRef]
- Teta, R.; Della Sala, G.; Glukhov, E.; Gerwick, L.; Gerwick, W.H.; Mangoni, A.; Costantino, V. Combined LC–MS/MS and molecular networking approach reveals new cyanotoxins from the 2014 cyanobacterial bloom in Green Lake, Seattle. Environ. Sci. Technol. 2015, 49, 14301–14310. [Google Scholar] [CrossRef]
- Ballot, A.; Sandvik, M.; Rundberget, T.; Botha, C.J.; Miles, C.O. Diversity of cyanobacteria and cyanotoxins in Hartbeespoort Dam, South Africa. Mar. Freshwater Res. 2014, 65, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Miles, C.O.; Sandvik, M.; Nonga, H.E.; Rundberget, T.; Wilkins, A.L.; Rise, F.; Ballot, A. Identification of microcystins in a Lake Victoria cyanobacterial bloom using LC–MS with thiol derivatization. Toxicon 2013, 70, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Namikoshi, M.; Yuan, M.; Sivonen, K.; Carmichael, W.W.; Rinehart, K.L.; Rouhiainen, L.; Sun, F.; Brittain, S.; Otsuki, A. Seven new microcystins possessing two l-glutamic acid units, isolated from Anabaena sp. strain 186. Chem. Res. Toxicol. 1998, 11, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Okello, W.; Portmann, C.; Erhard, M.; Gademann, K.; Kurmayer, R. Occurrence of microcystin-producing cyanobacteria in Ugandan freshwater habitats. Environ. Toxicol. 2010, 25, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, G.; Wang, D.; Gu, J.; Shen, Q.; Gross, S.S.; Wang, Y. Neutral loss of isocyanic acid in peptide CID spectra: A novel diagnostic marker for mass spectrometric identification of protein citrullination. J. Am. Soc. Mass Spectrom. 2009, 20, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Cunin, R.; Glansdorff, N.; Pierard, A.; Stalon, V. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 1986, 50, 314–352. [Google Scholar]
- Kotai, J. Instructions for Preparation of Modified Nutrient Solution Z8 for Algae; Norwegian Institute for Water Research: Oslo, Norway, 1972. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microcystin | Confidence | Neutral Formula a | No. N b | tR (min) c | Positive d m/z | +z | Δm (ppm) | Thiol-Reactive | No. CO2H e | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | [d-Asp3]MC-RR | confirmed | C48H73N13O12 | 13 | 2.81 | 512.7815 | 2 | −1.6 | yes | 2 |
| 4 | [d-Asp3]MC-LR | confirmed | C48H72N10O12 | 10 | 5.72 | 981.5419 | 1 | +1.6 | yes | 2 |
| 11 | [d-Asp3,Mser7]MC-RR | probable | C48H75N13O13 | 13 | 2.59 | 521.7879 | 2 | +0.5 | no | ND |
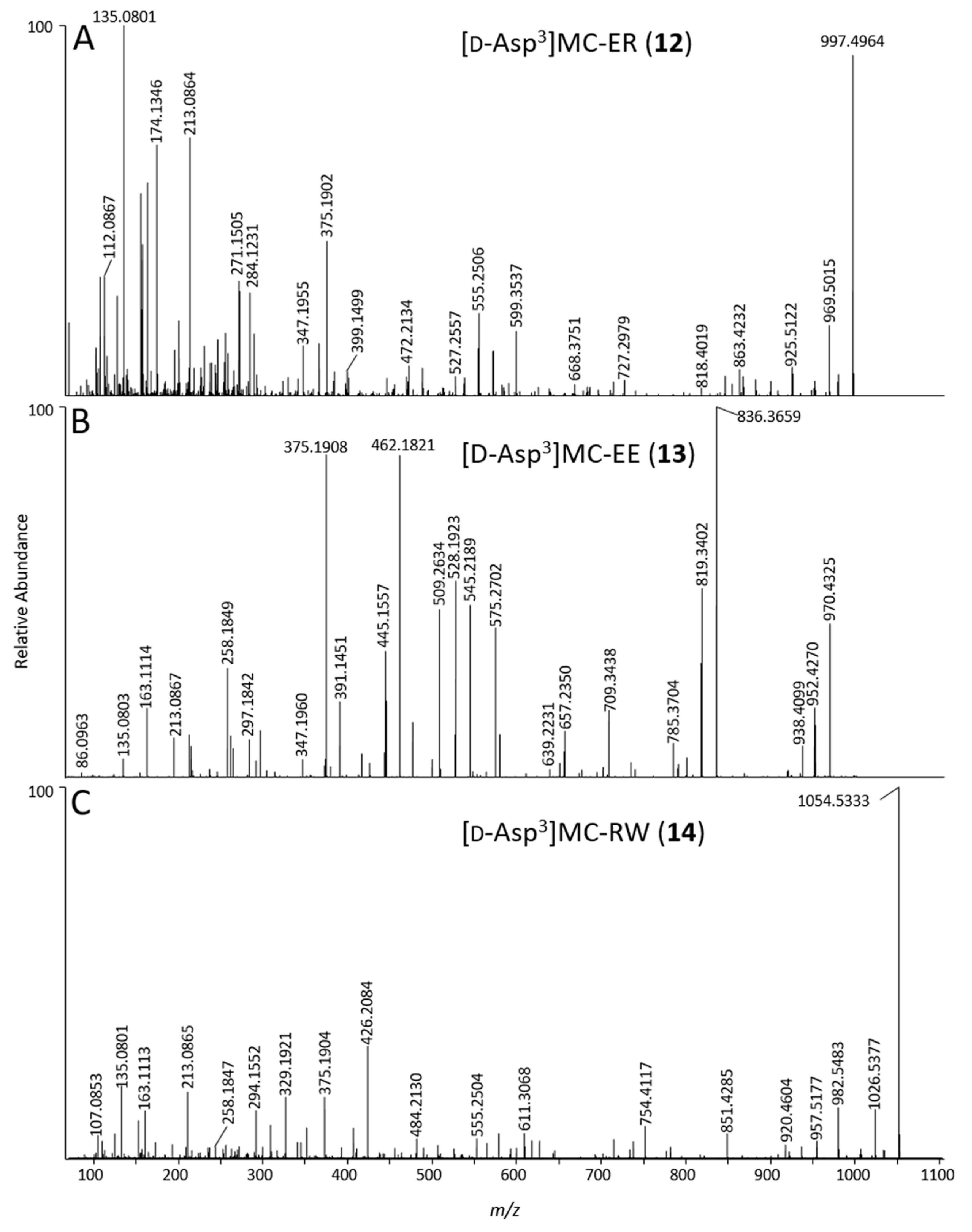
| 12 | [d-Asp3]MC-ER | probable | C47H68N10O14 | 10 | 3.63 | 997.4988 | 1 | −0.2 | yes | 3 |
| 13 | [d-Asp3]MC-EE | probable | C46H63N7O16 | 7 | 4.99 | 970.4413 | 1 | +0.9 | yes | 4 |
| 14 | [d-Asp3]MC-RW | probable | C53H71N11O12 | 11 | 10.01 | 1054.5387 | 1 | +2.9 | yes | ND |
| 15 | Sulfide conjugate of 1 | tentative | C93H145N21O33S | 21 | 9.85 f | 1059.0076 | 2 | −1.3 | no | ND |
| 16 | 15-sulfoxide | tentative | C93H145N21O34S | 21 | 6.50 g | 1067.0052 | 2 | −1.2 | no | ND |
| 17 | [d-Asp3]MC-RY | probable | C51H70N10O13 | 10 | 7.16 | 1031.5204 | 1 | +0.7 | yes | ND |
| 18 | [d-Asp3]MC-RF | probable | C51H70N10O12 | 10 | 9.94 | 1015.5246 | 1 | −0.2 | yes | ND |
| 19 | GSH-conjugate of 1 | confirmed | C58H90N16O18S | 16 | 2.05 | 666.3251 | 2 | +1.3 | no | ND |
| 20 | [d-Asp3]MC-RCit | probable | C48H72N12O13 | 12 | 3.44 | 1025.5431 | 1 | +1.6 | yes | ND |
| Fragment Ion Assignment | (6) | (5) | (4) | (12) |
|---|---|---|---|---|
| [M + H]+ | 995.6 | 981.5 | 981.5 | 997.5 |
| [M − NH3+ H]+ | 978.6 | 964.5 | 964.5 | 980.5 |
| [M − H2O +H]+ | 977.6 | 963.6 | 963.5 | 979.5 |
| [M − CO + H]+ | 967.6 | 953.6 | 953.6 | 969.5 |
| [M − 48 + H]+ | 946.5 | 932.5 | 932.5 | 948.4 |
| [Arg-Adda-Glu-res7-Ala-X2-NH2 + 2H]+ | 883.6 | 869.6 | 883.5 | 899.5 |
| [Arg-Adda-Glu-res7-Ala-X2 + H]+ | 866.6 | 852.5 | 866.6 | 882.5 |
| [res7-Ala-X2-res3-Arg-Adda + H]+ | 866.6 | 852.5 | 852.5 | 868.5 |
| [M−Addafrag. + H]+ | 861.5 | 847.5 | 847.5 | 863.4 |
| [M−Addafrag. − NH3 + H]+ | 844.5 | 830.4 | 830.4 | 846.4 |
| [Arg-Adda-Glu-res7-Ala- X2-CO + H]+ | 838.6 | 824.6 | 838.6 | 854.5 |
| [Ala-X2-res3-Arg-Adda − Addafrag. + H]+ | 783.5 | 783.5 | 769.6 | 785.4 |
| [Arg-Adda-Glu-res7-Ala + H]+ | 753.5 | 739.5 | 753.5 | 753.5 |
| [res3-Arg-Adda-Glu + H]+ | 728.5 | 728.5 | 714.4 | 714.4 |
| [res3-Arg-Adda-Glu − H2O + H]+ | 710.4 | 710.4 | 696.4 | 696.4 |
| [Arg-Adda-Glu-res7 + H]+ | 682.4 | 668.4 | 682.4 | 696.4 |
| [Glu-res7-Ala-X2-res3-Arg + H]+ | 682.4 | 668.4 | 668.4 | 684.4 |
| [Arg-Adda-Glu + H]+ | 599.4 | 599.4 | 599.4 | 599.4 |
| [Arg-Adda-Glu − NH3 + H]+ | 582.4 | 582.4 | 582.4 | 582.4 |
| [Arg-Adda-Glu − CO + H]+ | 571.4 | 571.4 | 571.4 | 571.4 |
| [res7-Ala-X2-res3-Arg-NH2 + 2H]+ | 570.4 | 556.4 | 556.4 | 572.3 |
| [res7-Ala-X2-res3-Arg + H]+ | 553.4 | 539.4 | 539.4 | 555.3 |
| [Ala-X2-res3-Arg + H]+ | 470.4 | 470.4 | 456.4 | 472.3 |
| [Ala-X2-res3-Arg − NH3 + H]+ | 453.3 | 453.3 | 439.3 | 455.3 |
| [Adda-Glu-res7 − Addafrag. − NH3 + H]+ | 375.3 | 361.2 | 375.3 | 375.3 |
| [res3-Arg-NH2 + H]+ | 303.2 | 303.2 | 289.2 | 289.2 |
| [res3-Arg + H]+ | 285.2 | 285.2 | ND | ND |
| Fragment Ion Assignment | (21) | (22) | (13) |
|---|---|---|---|
| [M + H]+ | 988 | 972 | 970.4 |
| [M − NH3 + H]+ | 971 | 955 | 953.4 |
| [M − H2O + H]+ | 970 | 954 | 952.5 |
| [M − CO + H]+ | 960 | 944 | 942.6 |
| [M − Addafrag + H]+ | 854 | 838 | 836.4 |
| [M − Addafrag − NH3 + H]+ | 837 | 821 | 819.4 |
| [M − Addafrag − H2O + H]+ | 836 | 820 | 818.5 |
| [Adda-Glu-Mdha-Ala-X2 − NH3+ H]+ | 693 | 693 | 709.3 |
| [M − Adda + H]+ | 675 | 659 | 657.3 |
| [M − Adda − H2O + H]+ | ND | ND | 639.2 |
| [Adda-Glu-Mdha-Ala − NH3 + H]+ | 580 | 580 | 580.3 |
| [Adda-Glu-Mdha-Ala-X2 − Addafrag − NH3+ H]+ | 559 | 559 | 575.2 |
| [Z4-Asp-X2-Ala-Mdha-NH2 + 2H]+ | 563 | 547 | 545.4 |
| [Z4-Asp-X2-Ala-Mdha + H]+ | 546 | 530 | 528.3 |
| [Adda-Glu-Mdha − NH3 + H]+ | 509 | 509 | 509.3 |
| [Z4-Asp-X2-Ala-NH2 + 2H]+ | 480 | 464 | 462.3 |
| [Z4-Asp-X2-Ala + H]+ | 463 | 447 | 445.2 |
| [Z4-Asp-X2 + H]+ | 392 | 376 | 374.3 |
| [Adda-Glu-Mdha − Addafrag − NH3 + H]+ | 375 | 375 | 375.3 |
| Fragment Ion Assignment | 17 | 18 | 14 |
|---|---|---|---|
| [M + H]+ | 1031 | 1015 | 1054.8 |
| [M − NH3 + H]+ | 1014 | 998 | 1037.7 |
| [M − H2O + H]+ | 1013 | 997 | 1036.7 |
| [M − CO + H]+ | 1003 | 987 | 1026.7 |
| [Z4-Adda-Glu-Mdha-Ala-Arg − NH3 + H]+ | 916 | 900 | 939.6 |
| [M − Addafrag + H]+ | 897 | 881 | 920.7 |
| [Adda-Glu-Mdha-Ala-Arg-Asp − NH3 + H]+ | 851 | 851 | 851.6 |
| [Ala-Arg-Asp-Z4-Adda + H]+ | 819 | 803 | 842.5 |
| [Adda-Glu-Mdha-Ala-Arg + H]+ | 754 | 754 | 754.6 |
| [Adda-Glu-Mdha-Ala-Arg − H2O + H]+ | 736 | 736 | 736.5 |
| [Adda-Glu-Mdha-Ala-Arg-Asp − Addafrag − NH3 + H]+ | 717 | 717 | 717.5 |
| [Mdha-Ala-Arg-Asp-Z4-NH2 + H]+ | 606 | 590 | 629.5 |
| [Mdha-Ala-Arg-Asp-Z4-NH2 − H2O + H]+ | 588 | 572 | 611.5 |
| [Mdha-Ala-Arg-Asp-Z4 − NH3 + H]+ | 572 | 556 | 595.3 |
| [Glu-Mdha-Ala-Arg-Asp + H]+ | 555 | ND | 555.4 |
| [Arg-Asp-Z4 + H]+ | 435 | 419 | 458.4 |
| [Mdha-Ala-Arg-Asp + H]+ | 426 | 426 | 426.4 |
| [Mdha-Ala-Arg-Asp − NH3 + H]+ | 409 | 409 | 409.3 |
| [Glu-Mdha-Ala-Arg − CO2H + H]+ | 395 | 395 | 395.3 |
| [Adda-Glu-Mdha − Addafrag − NH3 + H]+ | 375 | 375 | 375.3 |
| [Mdha-Ala-Arg + H]+ | 311 | 311 | 311.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallia, V.; Uhlig, S.; Rafuse, C.; Meija, J.; Miles, C.O. Novel Microcystins from Planktothrix prolifica NIVA-CYA 544 Identified by LC-MS/MS, Functional Group Derivatization and 15N-labeling. Mar. Drugs 2019, 17, 643. https://doi.org/10.3390/md17110643
Mallia V, Uhlig S, Rafuse C, Meija J, Miles CO. Novel Microcystins from Planktothrix prolifica NIVA-CYA 544 Identified by LC-MS/MS, Functional Group Derivatization and 15N-labeling. Marine Drugs. 2019; 17(11):643. https://doi.org/10.3390/md17110643
Chicago/Turabian StyleMallia, Vittoria, Silvio Uhlig, Cheryl Rafuse, Juris Meija, and Christopher O. Miles. 2019. "Novel Microcystins from Planktothrix prolifica NIVA-CYA 544 Identified by LC-MS/MS, Functional Group Derivatization and 15N-labeling" Marine Drugs 17, no. 11: 643. https://doi.org/10.3390/md17110643
APA StyleMallia, V., Uhlig, S., Rafuse, C., Meija, J., & Miles, C. O. (2019). Novel Microcystins from Planktothrix prolifica NIVA-CYA 544 Identified by LC-MS/MS, Functional Group Derivatization and 15N-labeling. Marine Drugs, 17(11), 643. https://doi.org/10.3390/md17110643