Chromone-Derived Polyketides from the Deep-Sea Fungus Diaporthe phaseolorum FS431


Abstract
:1. Introduction
2. Results and Discussion
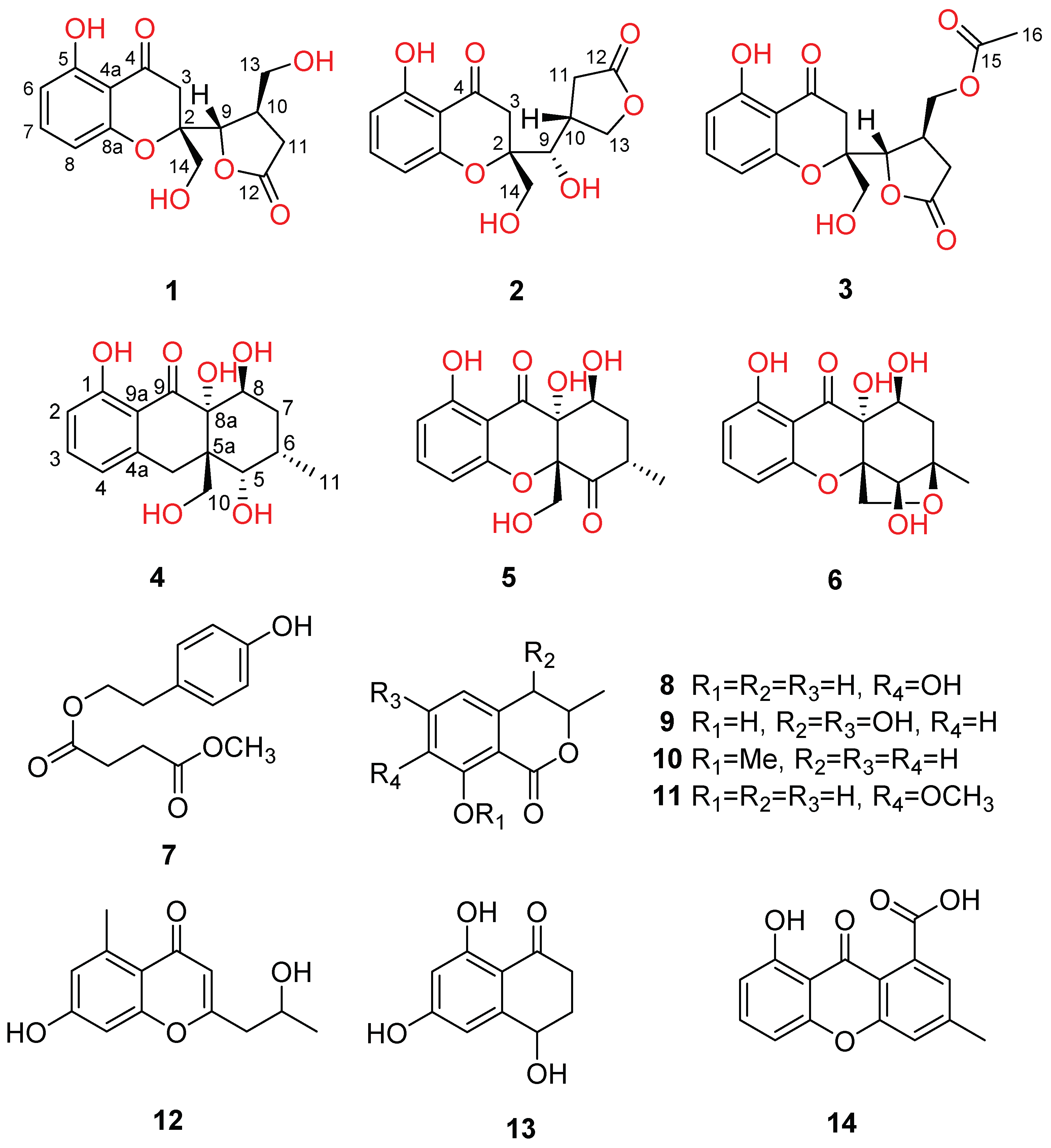
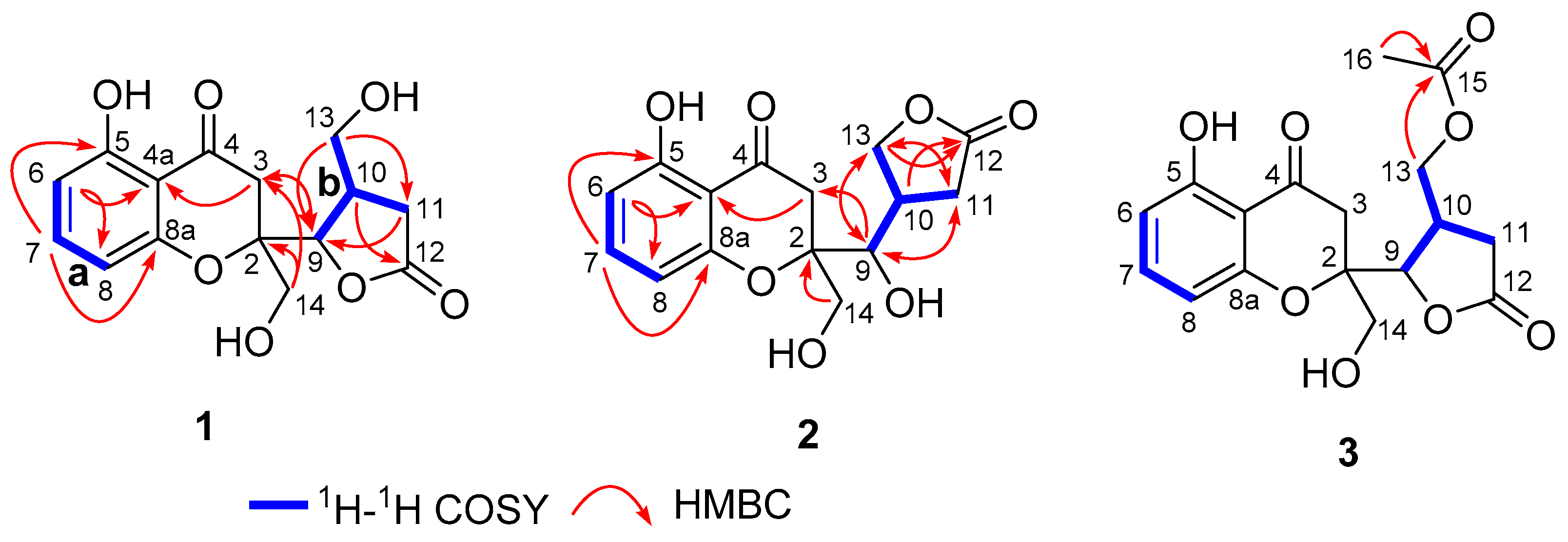
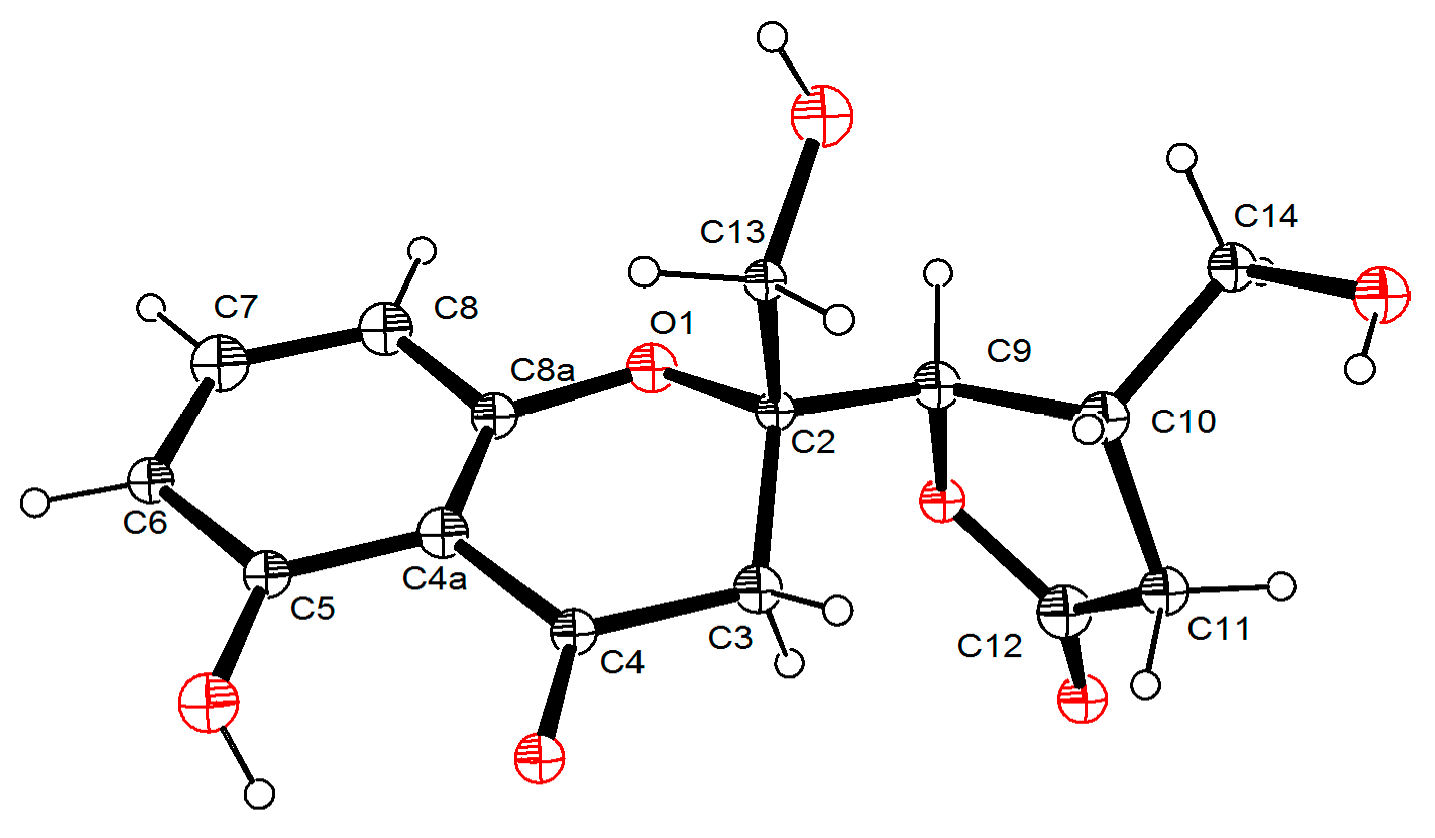
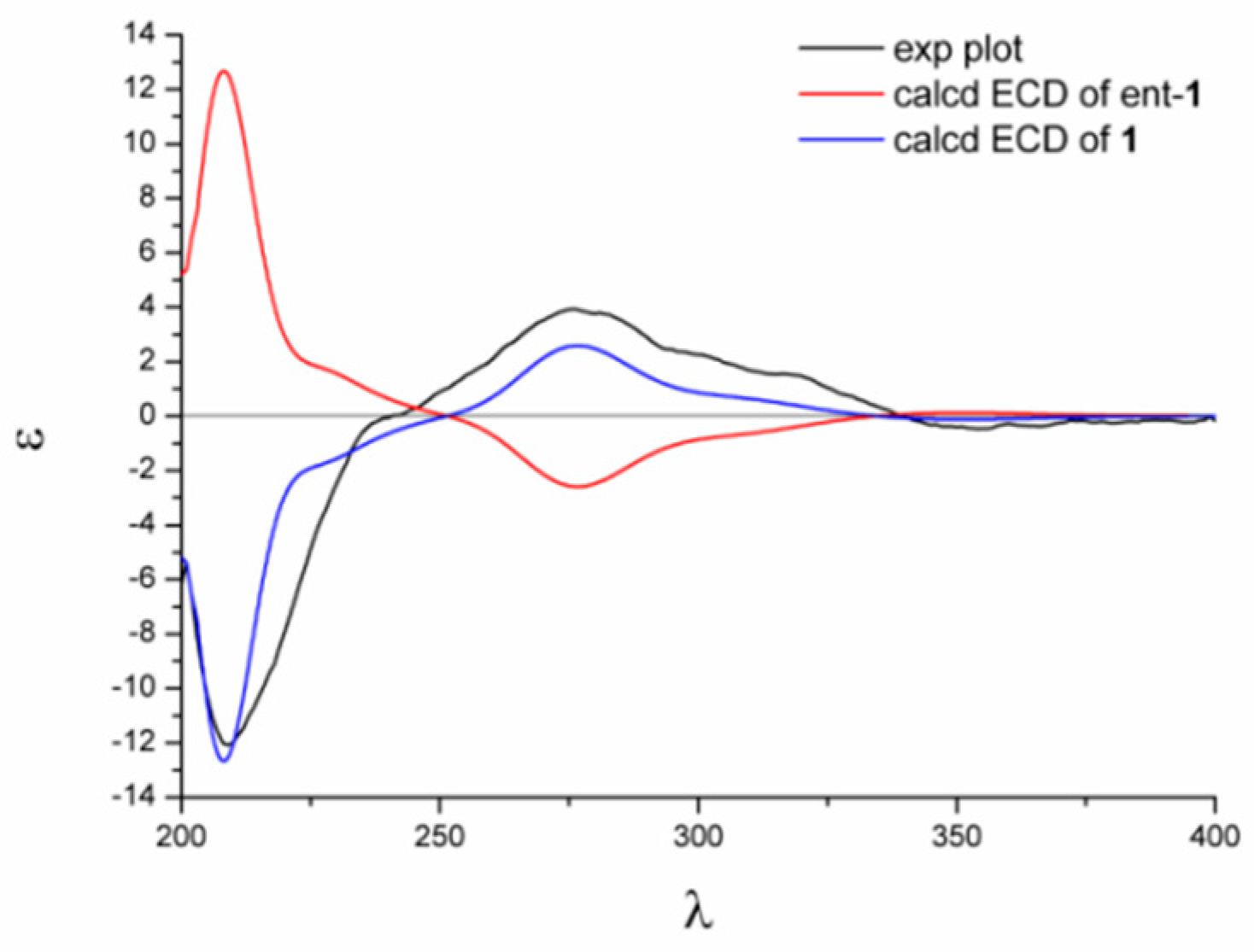
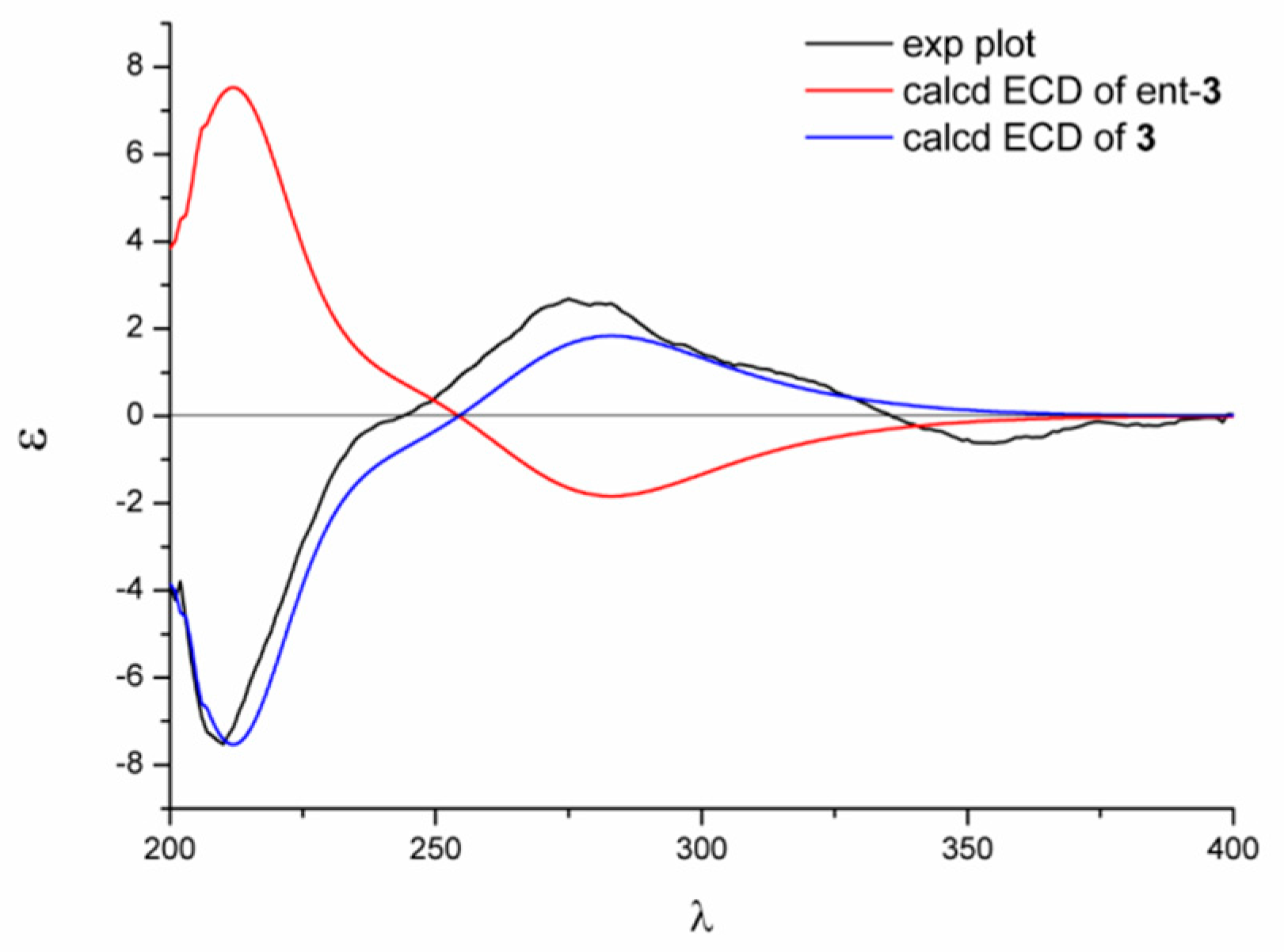
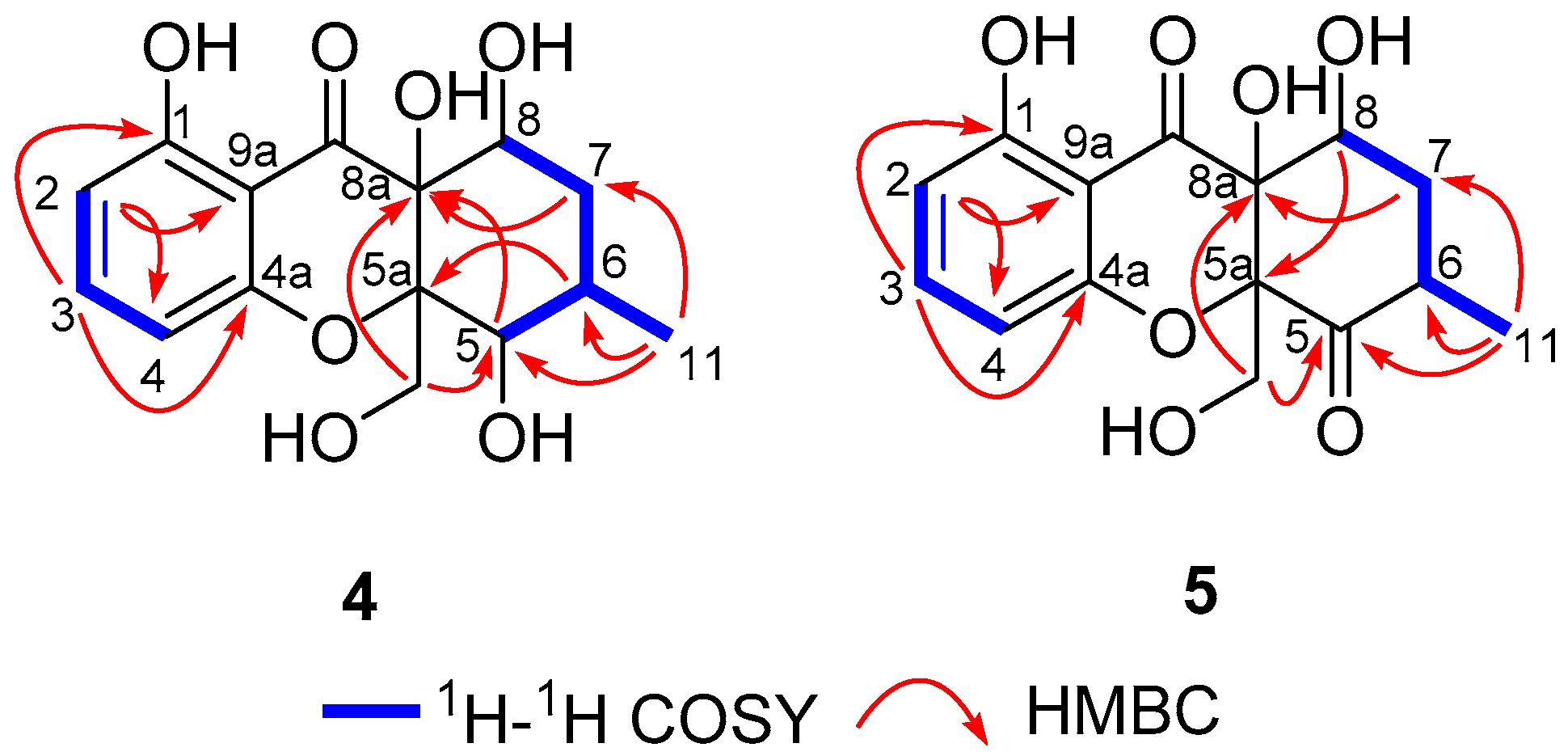
2.1. Structure Elucidation
2.2. Biological Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Isolation and Purification
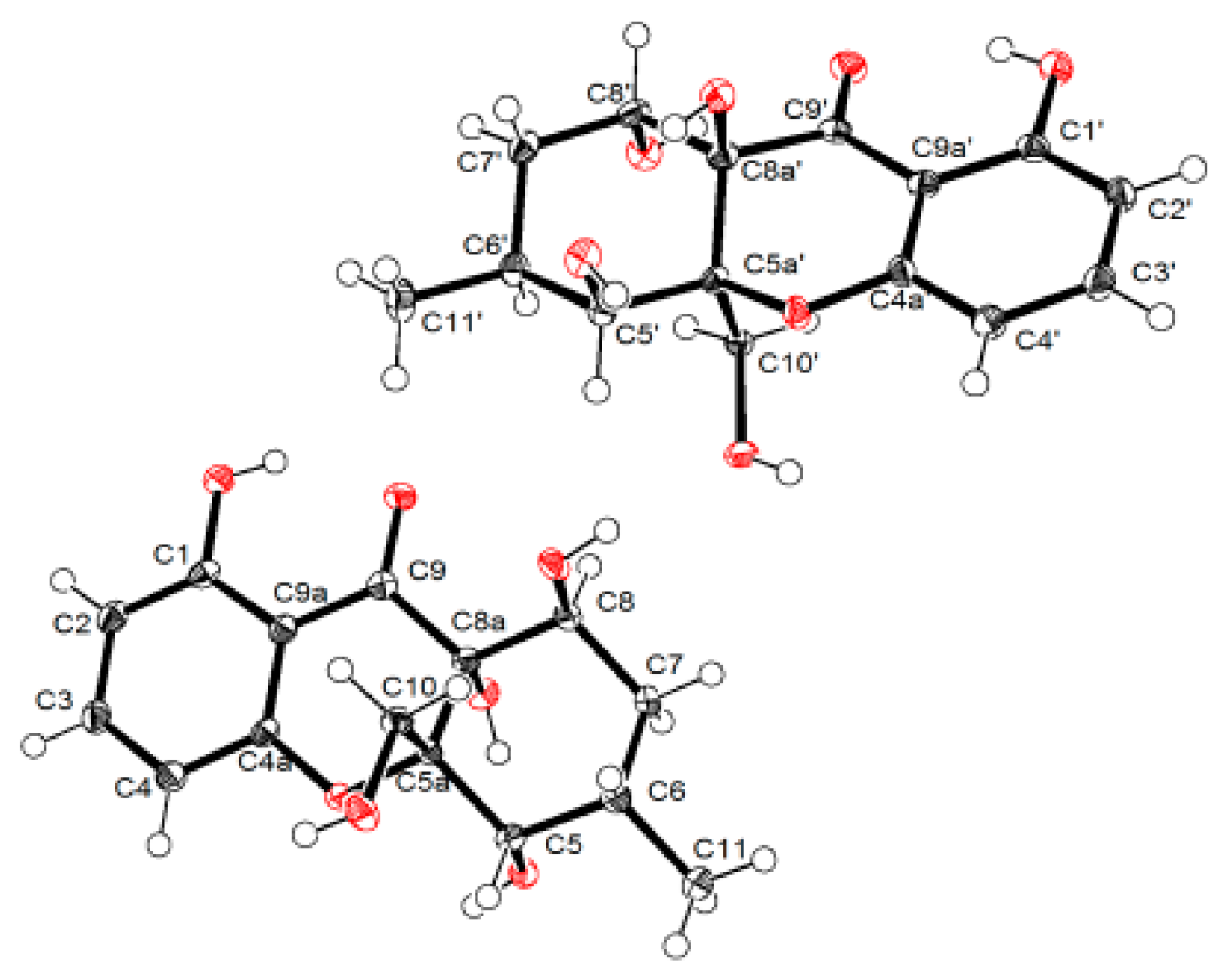
3.5. X-ray Crytallographic Data of Compounds 1 and 4
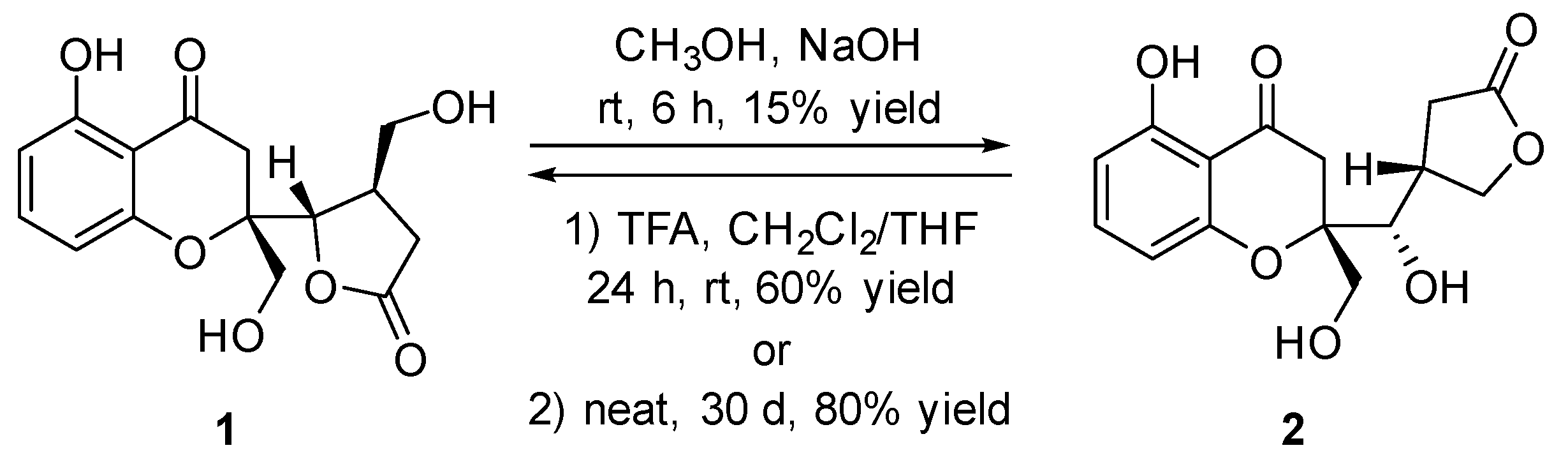
3.6. The Chemical Transformation Between Compounds 1 and 2
3.7. Cytotoxic Activity Assay
3.8. Details of ECD Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kijjoa, A.; Sawangwong, P. Drugs and cosmetics from the sea. Mar. Drugs 2004, 2, 73–82. [Google Scholar] [CrossRef]
- Bhadury, P.; Mohammad, B.T.; Wright, P.C. The current status of natural products from marine fungi and their potential as anti-infective agents. J. Ind. Microbiol. Biot. 2006, 33, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, Z.; Lin, X.; Lu, C.; Shen, Y. A new polyketide from Diaporthe sp. SXZ-19, an endophytic fungal strain of Camptotheca acuminate. Nat. Prod. Res. 2013, 27, 2100–2104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ondeyka, J.G.; Herath, K.B.; Guan, Z.; Collado, J.; Platas, G.; Pelaez, F.; Leavitt, P.S.; Gurnett, A.; Nare, B.; et al. Tenellones A and B from a Diaporthe sp.: Two highly substituted benzophenone inhibitors of parasite cGMP-dependent protein kinase activity. J. Nat. Prod. 2005, 68, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Andolfi, A.; Boari, A.; Evidente, M.; Cimmino, A.; Vurro, M.; Ash, G.; Evidente, A. Gulypyrones A and B and phomentrioloxins B and C produced by Diaporthe gulyae, a potential mycoherbicide for saffron thistle (Carthamus lanatus). J. Nat. Prod. 2015, 78, 623–629. [Google Scholar] [CrossRef]
- Zang, L.Y.; Wei, W.; Guo, Y.; Wang, T.; Jiao, R.H.; Ng, S.W.; Tan, R.X.; Ge, H.M. Sesquiterpenoids from the mangrove-derived endophytic fungus Diaporthe sp. J. Nat. Prod. 2012, 75, 1744–1749. [Google Scholar] [CrossRef]
- Yedukondalu, N.; Arora, P.; Wadhwa, B.; Malik, F.A.; Vishwakarma, R.A.; Gupta, V.K.; Riyazulhassan, S.; Ali, A. Diapolic acid A-B from an endophytic fungus, Diaporthe terebinthifolii depicting antimicrobial and cytotoxic activity. J. Antibiot. 2016, 70, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.F.; Wagenaar, M.M.; Singh, M.P.; Janso, J.E.; Clardy, J. The cytosporones, new octaketide antibiotics isolated from an endophytic fungus. Org. Lett. 2000, 2, 4043–4046. [Google Scholar] [CrossRef]
- Li, G.; Kusari, S.; Kusari, P.; Kayser, O.; Spiteller, M. Endophytic Diaporthe sp. LG23 produces a potent antibacterial tetracyclic triterpenoid. J. Nat. Prod. 2015, 78, 2128–2132. [Google Scholar] [CrossRef]
- Calcul, L.; Waterman, C.; Ma, W.S.; Lebar, M.D.; Harter, C.; Mutka, T.; Morton, L.; Maignan, P.; Van Olphen, A.; Kyle, D.E.; et al. Screening mangrove endophytic fungi for antimalarial natural products. Mar. Drugs 2013, 11, 5036–5050. [Google Scholar] [CrossRef]
- Mandavid, H.; Rodrigues, A.M.; Espindola, L.S.; Eparvier, V.; Stien, D. Secondary metabolites isolated from the amazonian endophytic fungus Diaporthe sp. SNB-GSS10. J. Nat. Prod. 2015, 78, 1735–1739. [Google Scholar] [CrossRef]
- Schloss, S.; Hackl, T.; Herz, C.; Lamy, E.; Koch, M.; Rohn, S.; Maul, R. Detection of a toxic methylated derivative of phomopsin A produced by the legume-infesting fungus Diaporthe toxica. J. Nat. Prod. 2017, 80, 1930–1934. [Google Scholar] [CrossRef]
- Hu, H.B.; Luo, Y.F.; Wang, P.; Wang, W.J.; Wu, J. Xanthone-derived polyketides from the thai mangrove endophytic fungus Phomopsis sp. xy21. Fitoterapia 2018, 131, 265–271. [Google Scholar] [CrossRef]
- Wang, Y.N.; Tian, L.; Hua, H.M.; Lu, X.; Sun, S.; Wu, H.H.; Pei, Y.H. Two new compounds from the broth of the marine fungus Penicillium griseofulvum Y19-07. J. Asian. Nat. Prod. Res. 2009, 11, 912–917. [Google Scholar] [CrossRef]
- Siddiqui, S.; Mahmood, T.; Siddiqui, B.S.; Faizi, S. Non-terpenoidal constituents from Azadirachta indica. Planta Med. 1988, 54, 457–459. [Google Scholar] [CrossRef]
- Devys, M.; Bousquet, J.F.; Kollmann, A.; Barbier, M. Dihydroisocoumarins and mycophenolic acid of the culture medium of the plant pathogenic fungus Septoria nodorum. Phytochemistry 1980, 19, 2221–2222. [Google Scholar] [CrossRef]
- Avantaggiato, G.; Solfrizzo, M.; Tosi, L.; Zazzerini, A.; Fanizzi, F.P.; Visconti, A. Isolation and characterization of phytotoxic compounds produced by Phomopsis helianthi. Nat. Toxins. 1999, 7, 119–127. [Google Scholar] [CrossRef]
- Glauser, G.; Gindro, K.; Fringeli, J.; De Joffrey, J.P.; Rudaz, S.; Wolfender, J.L. Differential analysis of mycoalexins in confrontation zones of grapevine fungal pathogens by ultrahigh pressure liquid chromatography/time-of-flight mass spectrometry and capillary nuclear magnetic resonance. J. Agric. Food Chem. 2009, 57, 1127–1134. [Google Scholar] [CrossRef]
- Mali, R.S.; Jagtap, P.G.; Patil, S.R.; Pawar, P.N. Novel AlCl3 catalysed syntheses of naturally occurring (±)8-hydroxy-3-rnethyl-3.4-dihydroisocoumarins. J. Chem. Soc. Chem. Comm. 1992, 0, 883–884. [Google Scholar] [CrossRef]
- Xu, J.; Kjer, J.; Sendker, J.; Wray, V.; Guan, H.S.; Edrada, R.A.; Lin, W.H.; Wu, J.; Proksch, P. Chromones from the endophytic fungus Pestalotiopsis sp. isolated from the chinese mangrove plant Rhizophora mucronata. J. Nat. Prod. 2009, 72, 662–665. [Google Scholar] [CrossRef]
- Venkatasubbaiah, P.; Chilton, W.S. Phytotoxins produced by Tubakia dryina. Mycopathologia 1992, 120, 33–37. [Google Scholar] [CrossRef]
- Krick, A.; Kehraus, S.; Gerhauser, C.; Klimo, K.; Nieger, M.; Maier, A.; Fiebig, H.H.; Atodiresei, I.; Raabe, G.; Fleischhauer, J.; et al. Potential cancer chemopreventive in vitro activities of monomeric xanthone derivatives from the marine algicolous fungus Monodictys putredinis. J. Nat. Prod. 2007, 70, 353–360. [Google Scholar] [CrossRef]
- Yang, B.; Tao, H.M.; Lin, X.P.; Wang, J.F.; Liao, S.R.; Dong, J.D.; Zhou, X.F.; Liu, Y.H. Prenylated indole alkaloids and chromone derivatives from the fungus Penicillium sp. SCSIO041218. Tetrahedron 2017, 74, 77–82. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Wu, P.; Xue, J.; Yao, L.; Xu, L.; Li, H.; Wei, X. Bisacremines E-G, three polycyclic dimeric acremines produced by acremonium persicinum SC0105. Org. Lett. 2015, 17, 4922–4925. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumloffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 b | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 2 | 84.9, C | 86.4, C | ||
| 3α | 3.15, d, (17.4) | 38.4, CH2 | 3.15, d, (17.4) | 39.4, CH2 |
| 3β | 3.03, d, (17.4) | 3.05, d, (17.4) | ||
| 4 | 198.1, C | 199.1, C | ||
| 4a | 108.3, C | 109.2, C | ||
| 5 | 162.7, C | 162.4, C | ||
| 6 | 6.43, d, (8.3) | 108.6, CH | 6.42, d, (8.3) | 108.2, CH |
| 7 | 7.38, t, (8.3) | 139.6, CH | 7.40, t, (8.3) | 139.0, CH |
| 8 | 6.43, d, (8.3) | 110.1, CH | 6.40, d, (8.3) | 108.2, CH |
| 8a | 160.7, C | 160.7, C | ||
| 9 | 4.65, d, (2.9) | 84.4, CH | 4.04, t, (5.3) | 73.1, CH |
| 10 | 2.93, m | 38.3, CH | 3.15, m | 38.2, CH |
| 11α | 2.83, dd, (18.2, 10.1) | 32.0, CH2 | 2.66, dd, (17.3, 10.2) | 30.1, CH2 |
| 11β | 2.38, dd, (18.2, 3.1) | 2.50, dd (17.3, 8.7) | ||
| 12 | 179.0, C | 177.1, C | ||
| 13α | 3.59, m | 64.6, CH2 | 4.51, t, (9.0) | 71.5, CH2 |
| 13β | 4.08, t, (9.0) | |||
| 14 | 3.78, m | 62.9, CH2 | 3.91, br s, | 63.4, CH2 |
| No. | 3 | No. | 3 | ||
|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | ||
| 2 | 84.7, C | 9 | 4.66, d, (3.2) | 83.0, CH | |
| 3α | 3.14, d, (17.4) | 38.1, CH2 | 10 | 3.27, m | 35.1, CH |
| 3β | 3.03, d, (17.4) | 11α | 2.89, dd, (18.2, 10.2) | 31.7, CH2 | |
| 4 | 197.8, C | 11β | 2.42, dd, (18.2, 3.7) | ||
| 4a | 108.1, C | 12 | 175.9, C | ||
| 5 | 162.4, C | 13 | 4.20, d, (6.0) | 66.3, CH2 | |
| 6 | 6.44, d, (8.3) | 108.3, CH | 14 | 3.89, d, (3.0) | 63.0, CH2 |
| 7 | 7.41, t, (8.3) | 139.2, CH | 15 | 171.0, C | |
| 8 | 6.44, d, (8.3) | 109.6, CH | 16 | 2.02, s | 20.7, CH3 |
| 8a | 160.4, C | ||||
| No. | 4 a | 5 b | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 163.4, C | 163.6, C | ||
| 2 | 6.46, dd, (8.3, 0.9) | 109.7, CH | 6.51, dd, (8.3, 0.9) | 110.4, CH |
| 3 | 7.41, t, (8.3) | 139.0, CH | 7.46, t, (8.3) | 139.6, CH |
| 4 | 6.57, dd, (8.3, 0.9) | 109.5, CH | 6.65, dd, (8.3, 0.9) | 109.9, CH |
| 4a | 160.3, C | 159.0, C | ||
| 5 | 4.25, m | 74.5, CH | 207.1, C | |
| 5a | 76.4, C | 93.6, C | ||
| 6 | 2.22, m | 29.2, CH | 3.01, m | 38.1, CH |
| 7α | 2.04, m | 32.0, CH2 | 2.11, m | 39.2, CH2 |
| 7β | 1.57, m | |||
| 8 | 4.38, t, (2.9) | 68.5, CH | 4.51, t, (2.7) | 67.0, CH |
| 8a | 85.4, C | 79.2, C | ||
| 9 | 196.2, C | 196.2, C | ||
| 9a | 108.6, C | 107.9, C | ||
| 10α | 4.24, d, (13.5) | 60.5, CH2 | 3.86, d, (13.2) | 62.5, CH2 |
| 10β | 3.77, d, (13.5) | 4.84, d, (13.2) | ||
| 11 | 1.11, d, (6.7) | 18.1, CH3 | 1.11, s | 14.4, CH3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Liu, Z.-M.; Chen, Y.-C.; Tan, H.-B.; Li, S.-N.; Li, H.-H.; Gao, X.-X.; Liu, H.-X.; Zhang, W.-M. Chromone-Derived Polyketides from the Deep-Sea Fungus Diaporthe phaseolorum FS431. Mar. Drugs 2019, 17, 182. https://doi.org/10.3390/md17030182
Guo H, Liu Z-M, Chen Y-C, Tan H-B, Li S-N, Li H-H, Gao X-X, Liu H-X, Zhang W-M. Chromone-Derived Polyketides from the Deep-Sea Fungus Diaporthe phaseolorum FS431. Marine Drugs. 2019; 17(3):182. https://doi.org/10.3390/md17030182
Chicago/Turabian StyleGuo, Heng, Zhao-Ming Liu, Yu-Chan Chen, Hai-Bo Tan, Sai-Ni Li, Hao-Hua Li, Xiao-Xia Gao, Hong-Xin Liu, and Wei-Min Zhang. 2019. "Chromone-Derived Polyketides from the Deep-Sea Fungus Diaporthe phaseolorum FS431" Marine Drugs 17, no. 3: 182. https://doi.org/10.3390/md17030182
APA StyleGuo, H., Liu, Z. -M., Chen, Y. -C., Tan, H. -B., Li, S. -N., Li, H. -H., Gao, X. -X., Liu, H. -X., & Zhang, W. -M. (2019). Chromone-Derived Polyketides from the Deep-Sea Fungus Diaporthe phaseolorum FS431. Marine Drugs, 17(3), 182. https://doi.org/10.3390/md17030182