Total Synthesis of Mycalisine B


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
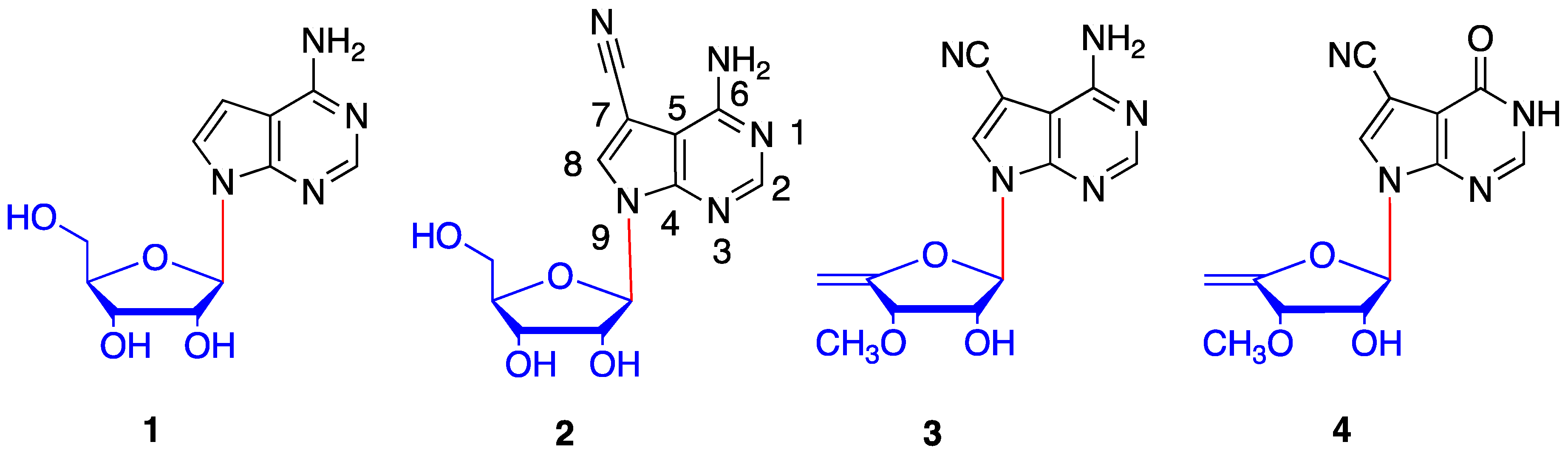
1. Introduction
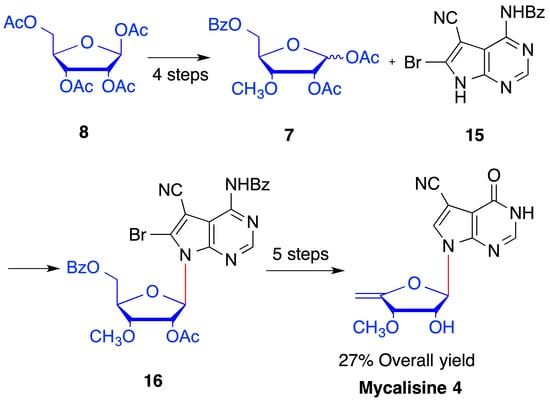
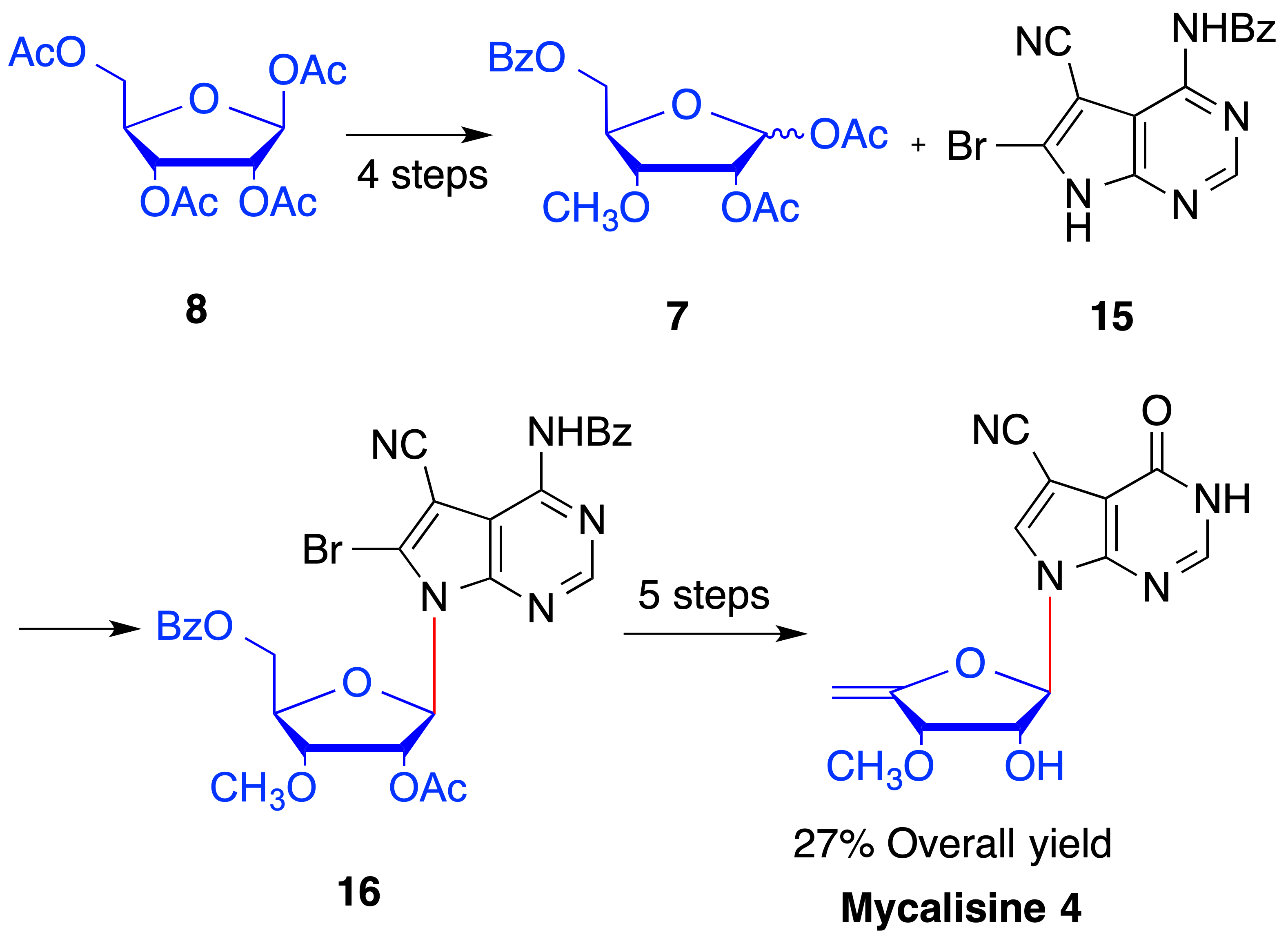
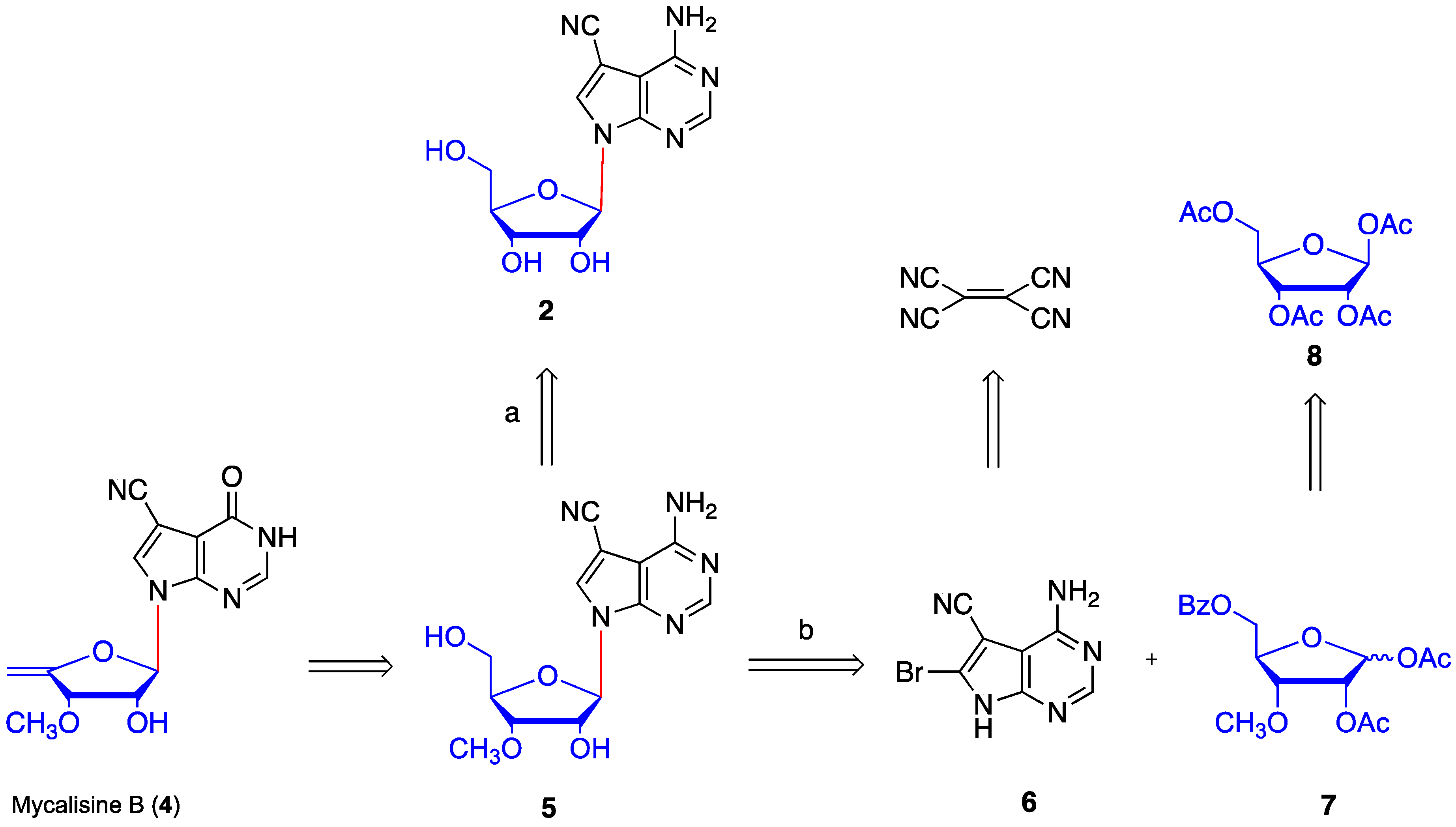
2. Results and Discussion
3. Conclusions
4. Materials and Methods
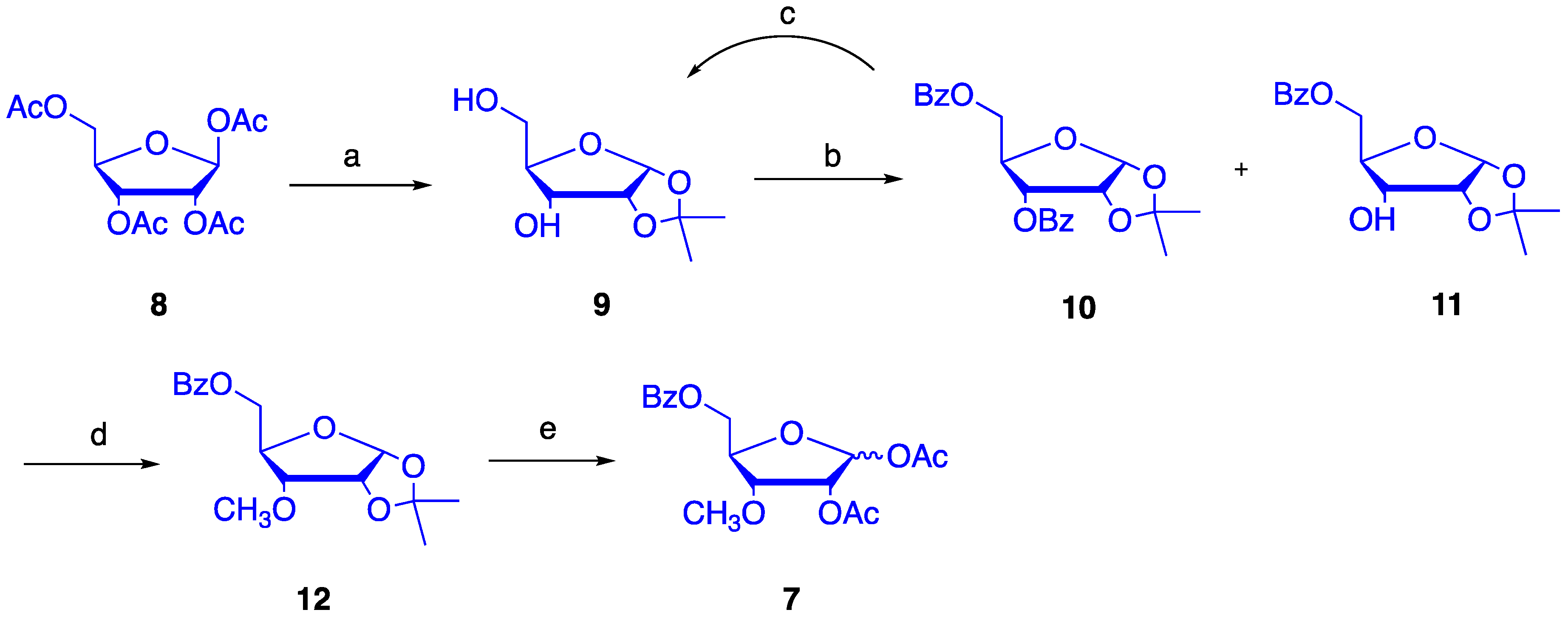
4.1. Synthesis of 1,2-O-isopropylidene-α-d-ribofuranose (9)
4.2. Synthesis of 1,2-O-isopropylidene-5-O-benzoyl-α-d-ribofuranose (11)
4.3. Synthesis of 1,2-O-isopropylidene-3-O-methyl-5-O-benzoyl-α-d-ribofuranose (12)
4.4. Synthesis of 1,2-O-diacetyl-3-O-methyl-5-O-benzoyl-d-ribofuranose (7)
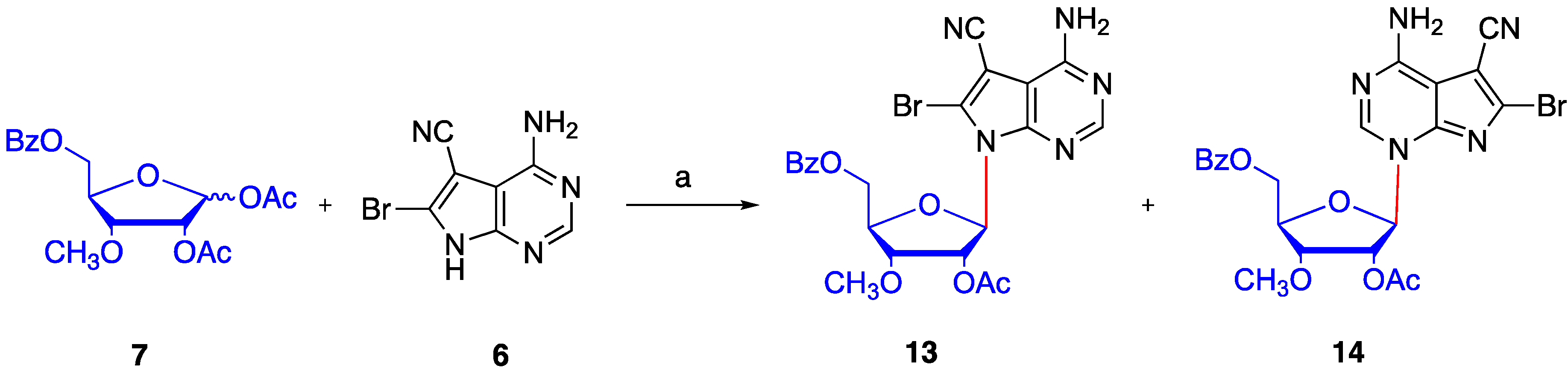
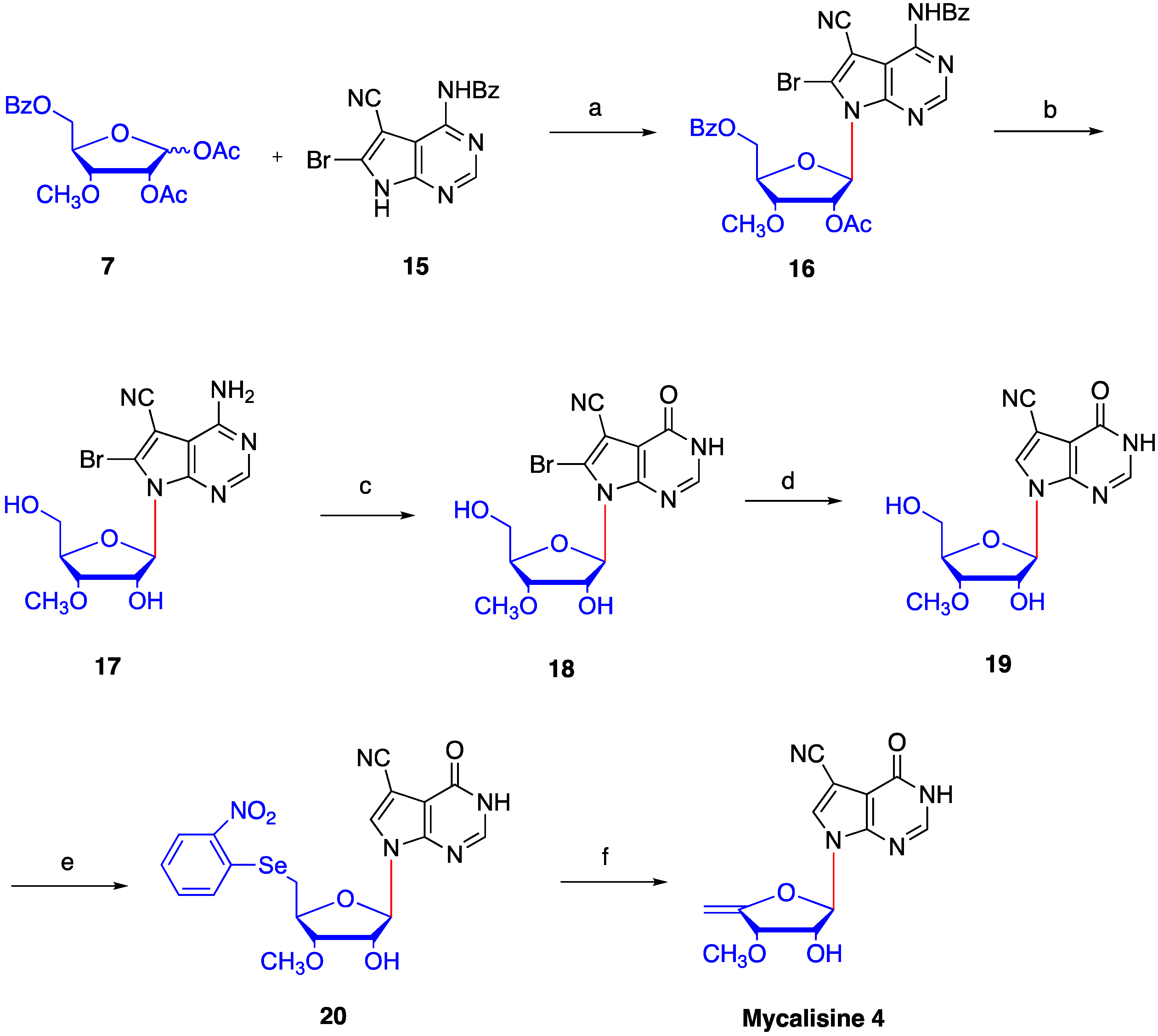
4.5. Synthesis of N4-benzoyl-5-cyano-6-bromo-7-(2′-O-acetyl -3′-O-methyl-5′-O-benzoyl-β-d- ribofuranosyl) -7H-pyrrolo[2,3-d] pyrimidine (16)
4.6. Synthesis of 5-cyano-6-bromo-7-(3′-O-methyl-β-d-ribofuranosyl)-7H-pyrrolo[2,3-d] pyrimidine (17)
4.7. 4-carbonyl-5-cyano-6-bromo-7-(3′-O-methyl-β-d-ribofuranosyl)-7H-pyrrolo[2,3-d] pyrimidine (18)
4.8. Synthesis of 4-carbonyl-5-cyano-7-(3′-O-methyl-β-d-ribofuranosyl)-7H-pyrrolo[2,3-d] pyrimidine (19)
4.9. Synthesis of 4-carbonyl-5-cyano-7-(3′-O-methyl-5′-O-(2-nitrophenyl) selanyl -β-d-ribofuranosyl)-7H- pyrrolo [2,3-d] pyrimidine (20)
4.10. Synthesis of mycalisine B (4)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Miller, J.H.; Field, J.J.; Kanakkanthara, A.; Owen, J.G.; Singh, A.J.; Northcote, P.T. Marine Invertebrate Natural Products that Target Microtubules. J. Nat. Prod. 2018, 81, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Farabegoli, F.; Blanco, L.; Rodriguez, L.P.; Manuel Vieites, J.; Garcia Cabado, A. Phycotoxins in Marine Shellfish: Origin, Occurrence and Effects on Humans. Mar. Drugs 2018, 16, 188. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, V.; Venkatesan, M.; Ramachandran, S.; Sundaresan, U. Bioactive Peptides from Marine Ascidians and Future Drug Development-A Review. Int. J. Pept. Res. Ther. 2018, 24, 13–18. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, M.; Chen, J.; Wang, H.; Tenney, K.; Crews, P. Bioactive Secondary Metabolites from the Marine Sponge Genus Agelas. Mar. Drugs 2017, 15, 351. [Google Scholar] [CrossRef] [PubMed]
- Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine Sponge Natural Products with Anticancer Potential: An Updated Review. Mar. Drugs 2017, 15, 310. [Google Scholar] [CrossRef] [PubMed]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine Sponge Derived Natural Products between 2001 and 2010: Trends and Opportunities for Discovery of Bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Fusetani, N.; Matsunaga, S.; Hashimoto, K. Bioactive marine metabolites IX. Mycalisines A and B, novel nucleosides which inhibit cell division of fertilized starfish eggs, from the marine sponge mycale sp. Tetrahedron Lett. 1985, 26, 3483–3486. [Google Scholar] [CrossRef]
- Anzai, K.; Nakamura, G.; Suzuki, S. A new antibiotic, tubercidin. J. Antibiot. 1957, 10, 201–204. [Google Scholar] [PubMed]
- Nishimura, H.; Katagiri, K.; Sato, K.; Mayama, M.; Shimaoka, N. Toyocamycin, a new anti-candida antibiotics. J. Antibiot. 1956, 9, 60–62. [Google Scholar] [PubMed]
- Chen, S.; Kinney, W.A.; Van Lanen, S. Nature’s combinatorial biosynthesis and recently engineered production of nucleoside antibiotics in Streptomyces. World J. Microbiol. Biotechnol. 2017, 33, 66. [Google Scholar] [CrossRef] [PubMed]
- Mulamoottil, V.A. Tubercidin and Related Analogues: An Inspiration for 50 years in Drug Discovery. Curr. Org. Chem. 2016, 20, 830–838. [Google Scholar] [CrossRef]
- McCarty, R.M.; Bandarian, V. Biosynthesis of pyrrolopyrimidines. Bioorg. Chem. 2012, 43, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snasel, J.; Naus, P.; Dostal, J.; Hnizda, A.; Fanfrlik, J.; Brynda, J.; Bourderioux, A.; Dusek, M.; Dvorakova, H.; Stolarikova, J.; et al. Structural Basis for Inhibition of Mycobacterial and Human Adenosine Kinase by 7-Substituted 7-(Het)aryl-7-deazaadenine Ribonucleosides. J. Med. Chem. 2014, 57, 8268–8279. [Google Scholar] [CrossRef] [PubMed]
- Ingale, S.A.; Pujari, S.S.; Sirivolu, V.R.; Ding, P.; Xiong, H.; Mei, H.; Seela, F. 7-Deazapurine and 8-Aza-7-deazapurine Nucleoside and Oligonucleotide Pyrene “Click” Conjugates: Synthesis, Nucleobase Controlled Fluorescence Quenching, and Duplex Stability. J. Org. Chem. 2012, 77, 188–199. [Google Scholar] [CrossRef]
- Minakawa, N.; Kawano, Y.; Murata, S.; Inoue, N.; Matsuda, A. Oligodeoxynucleotides containing 3-bromo-3-deazaadenine and 7-bromo-7-deazaadenine 2′-deoxynucleosides as chemical probes to investigate DNA-protein interactions. Chembiochem 2008, 9, 464–470. [Google Scholar] [CrossRef]
- Perlikova, P.; Hocek, M. Pyrrolo 2,3-d pyrimidine (7-deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med. Res. Rev. 2017, 37, 1429–1460. [Google Scholar] [CrossRef] [PubMed]
- Sheid, B.; Lerner, L.M.; Gaetjens, E. Antiproliferative effects of 4′,5′-unsaturated adenine nucleosides on leukemia L 1210 cells-invitro. Experientia 1989, 45, 729–730. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.R.; Jarvi, E.T.; Matthews, D.P.; Edwards, M.L.; Prakash, N.J.; Bowlin, T.L.; Mehdi, S.; Sunkara, P.S.; Bey, P. 4′,5′-Unsaturated 5′-fluoroadenosine nucleosides—Potent mechanism-based inhibitors of S-adenosyl-l-homocysteine hydrolase. J. Am. Chem. Soc. 1989, 111, 1127–1128. [Google Scholar] [CrossRef]
- Lamberth, C. The chemistry of 4′,5′-unsaturated nucleosides. A review. Org. Prep. Procedia Int. 1999, 31, 379–397. [Google Scholar] [CrossRef]
- Ding, H.; Li, C.; Dong, X.; Cao, B.; Zhang, N.; Hong, S.; Xiao, Q. First Total Synthesis of Marine Natural Nucleoside Kipukasin D. Chin. J. Org. Chem. 2018, 38, 3351–3355. [Google Scholar] [CrossRef]
- Li, C.; Ding, H.; Ruan, Z.; Zhou, Y.; Xiao, Q. First total synthesis of kipukasin A. Beilstein J. Org. Chem. 2017, 13, 855–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Li, W.; Ruan, Z.; Yang, R.; Mao, Z.; Xiao, Q.; Wu, J. Concise total synthesis of two marine natural nucleosides: Trachycladines A and B. Beilstein J. Org. Chem. 2014, 10, 1681–1685. [Google Scholar] [CrossRef]
- Sun, J.; Dou, Y.; Ding, H.; Yang, R.; Sun, Q.; Xiao, Q. First Total Synthesis of a Naturally Occurring Iodinated 5′-Deoxyxylofuranosyl Marine Nucleoside. Mar. Drugs 2012, 10, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.X.; Da, L.C.; Yang, R.C.; Cao, B.P.; Sun, Q.; Xiao, Q. First total synthesis of a naturally occurring nucleoside disulfide: 9-(5′-Deoxy-5′-thio-beta-D-xylofuranosyl)adenine disulfide. Chin. Chem. Lett. 2012, 23, 996–998. [Google Scholar] [CrossRef]
- Cao, B.; Ding, H.; Yang, R.; Wang, X.; Xiao, Q. Total Synthesis of a Marine Alkaloid-Rigidin E. Mar. Drugs 2012, 10, 1412–1421. [Google Scholar] [CrossRef]
- Song, Y.; Ding, H.; Dou, Y.; Yang, R.; Sun, Q.; Xiao, Q.; Ju, Y. Efficient and Practical Synthesis of 5′-Deoxytubercidin and Its Analogues via Vorbruggen Glycosylation. Synth. Stuttg. 2011, 1442–1446. [Google Scholar] [CrossRef]
- Dou, Y.-H.; Ding, H.-X.; Yang, R.-C.; Li, W.; Xiao, Q. A total synthesis of mycalisine A. Chin. Chem. Lett. 2013, 24, 379–382. [Google Scholar] [CrossRef]
- Meade, E.A.; Krawczyk, S.H.; Townsend, L.B. A total synthesis of the naturally-occurring pyrrolo[2,3-d] pyrimidine nucleoside, mycalisine-A. Tetrahedron Lett. 1988, 29, 4073–4076. [Google Scholar] [CrossRef]
- Robins, M.J.; Naik, S.R.; Lee, A.S. Nucleic acid related compounds. 12. The facile and high-yield stannous chloride catalyzed monomethylation of the cis-glycol system of nucleosides by diazomethane. J. Org. Chem. 1974, 39, 1891–1899. [Google Scholar] [CrossRef]
- More, J.D.; Campbell, M.G. Reaction of acetylated carbohydrates with trimethylaluminum: Concise synthesis of 1,2-O-isopropylidene d-ribofuranose. Tetrahedron Lett. 2009, 50, 2617–2619. [Google Scholar] [CrossRef]
- Houston, T.A.; Koreeda, M. Iodine-promoted ribosylation leads to a facile acetonide-forming reaction. Carbohydr. Res. 2009, 344, 2240–2244. [Google Scholar] [CrossRef] [PubMed]
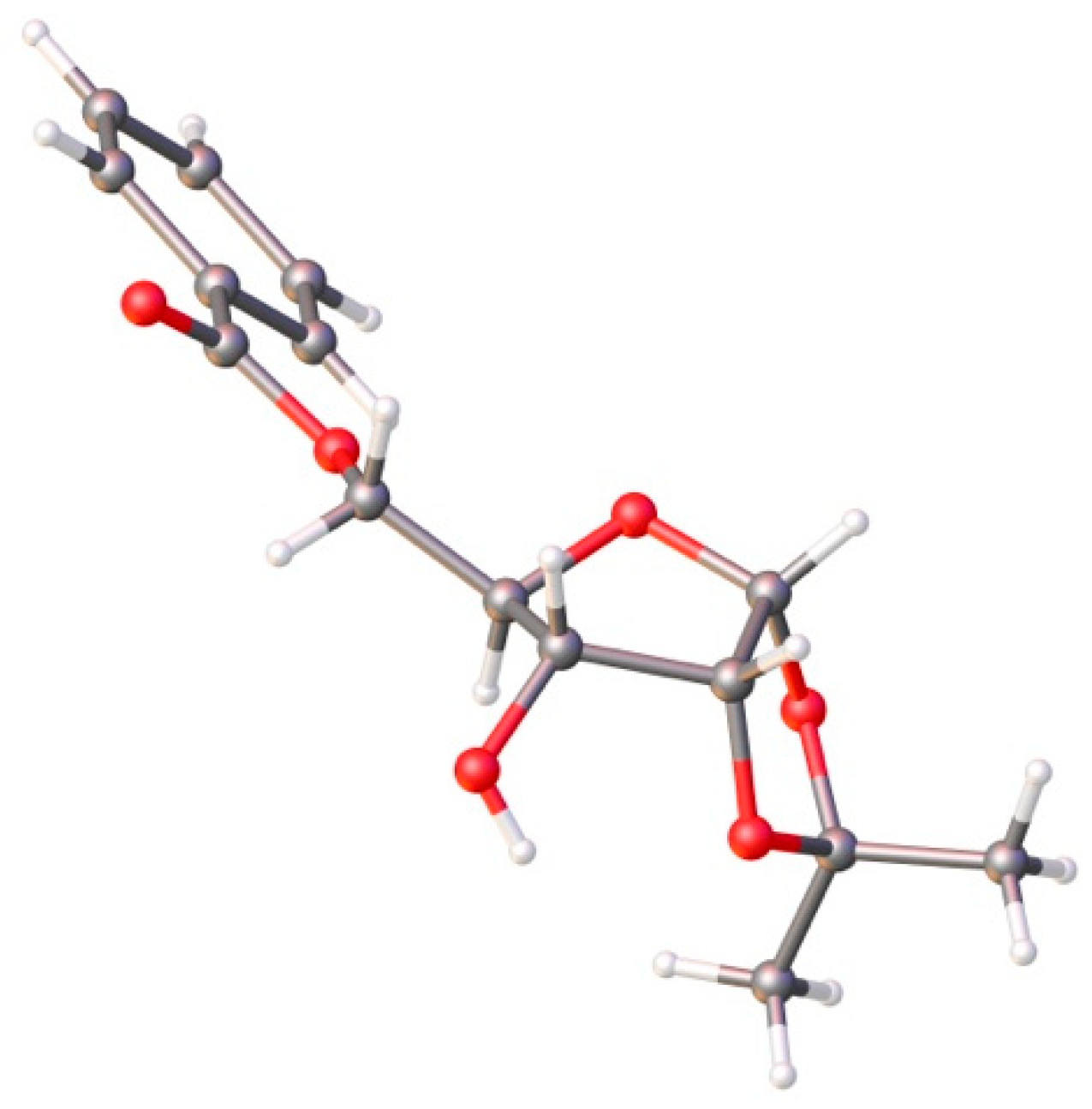
- Crystallographic Data for Ribose 11: C15H18O6, M = 294.29, Crystal Dimensions 0.3 × 0.28 × 0.25 mm, Colorless Block, Space Group, P21. CCDC 1901368 Contains the Supplementary Crystallographic Data. The Data Can Be Obtained Free of Charge from The Cambridge Crystallographic Date Centre via. Available online: www.ccdc.cam.ac.uk/date_request/cif (accessed on 13 April 2019).
- Ren, B.; Wang, M.; Liu, J.; Ge, J.; Zhang, X.; Dong, H. Zemplen transesterification: A name reaction that has misled us for 90 years. Green Chem. 2015, 17, 1390–1394. [Google Scholar] [CrossRef]
- Gibard, C. Silver(I) Oxide. Synlett 2014, 25, 2375–2376. [Google Scholar] [CrossRef] [Green Version]
- Seela, F.; Peng, X.H. Progress in 7-deazapurine (pyrrolo[2,3-d]pyrimidine) ribonucleoside synthesis. Curr. Top. Med. Chem. 2006, 6, 867–892. [Google Scholar] [CrossRef]
- Vorbruggen, H.; Krolikiewicz, K.; Bennua, B. Nucleoside syntheses XXII1) nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalysts. Chem. Ber.-Recl. 1981, 114, 1234–1255. [Google Scholar] [CrossRef]
- Vorbruggen, H.; Hofle, G. Nucleoside syntheses XXIII1) on the mechanism of nucleoside synthesis. Chem. Ber.-Recl. 1981, 114, 1256–1268. [Google Scholar] [CrossRef]
- Dong, X.; Tang, J.; Hu, C.; Bai, J.; Ding, H.; Xiao, Q. An Expeditious Total Synthesis of 5′-Deoxy-toyocamycin and 5′-Deoxysangivamycin. Molecules 2019, 24, 737. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Kwon, S.H.; Bae, I.H.; Kim, B.M. Selectivity between N-1 and N-7 nucleosides: Regioselective synthesis of BMK-Y101, a potent CDK7 and 9 inhibitor. Tetrahedron Lett. 2013, 54, 5484–5488. [Google Scholar] [CrossRef]
- Leonard, P.; Ingale, S.A.; Ding, P.; Ming, X.; Seela, F. Studies On The Glycosylation Of Pyrrolo 2,3-d Pyrimidines With 1-O-Acetyl-2,3,5-Tri-O-Benzoyl-Beta-d-Ribofuranose: The Formation Of Regioisomers During Toyocamycin And 7-Deazainosine Syntheses. Nucleosides Nucleotides Nucleic Acids 2009, 28, 678–694. [Google Scholar] [CrossRef]
- Ciliberti, N.; Durini, E.; Manfredini, S.; Vertuani, S. 7-Deazainosine Derivatives: Synthesis and Characterization of 7- and 7,8-Substituted Pyrrolo[2,3-d]Pyrimidine Ribonucleosides. Nucleosides Nucleotides Nucleic Acids 2008, 27, 525–533. [Google Scholar] [CrossRef]
- Grieco, P.A.; Gilman, S.; Nishizawa, M. Organoselenium chemistry. A facile one-step synthesis of alkyl aryl selenides from alcohols. J. Org. Chem. 1976, 41, 1485–1486. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, H.; Ruan, Z.; Kou, P.; Dong, X.; Bai, J.; Xiao, Q. Total Synthesis of Mycalisine B. Mar. Drugs 2019, 17, 226. https://doi.org/10.3390/md17040226
Ding H, Ruan Z, Kou P, Dong X, Bai J, Xiao Q. Total Synthesis of Mycalisine B. Marine Drugs. 2019; 17(4):226. https://doi.org/10.3390/md17040226
Chicago/Turabian StyleDing, Haixin, Zhizhong Ruan, Peihao Kou, Xiangyou Dong, Jiang Bai, and Qiang Xiao. 2019. "Total Synthesis of Mycalisine B" Marine Drugs 17, no. 4: 226. https://doi.org/10.3390/md17040226
APA StyleDing, H., Ruan, Z., Kou, P., Dong, X., Bai, J., & Xiao, Q. (2019). Total Synthesis of Mycalisine B. Marine Drugs, 17(4), 226. https://doi.org/10.3390/md17040226