Synthesis and Preliminary Biological Evaluation of Two Fluoroolefin Analogs of Largazole Inspired by the Structural Similarity of the Side Chain Unit in Psammaplin A

Abstract
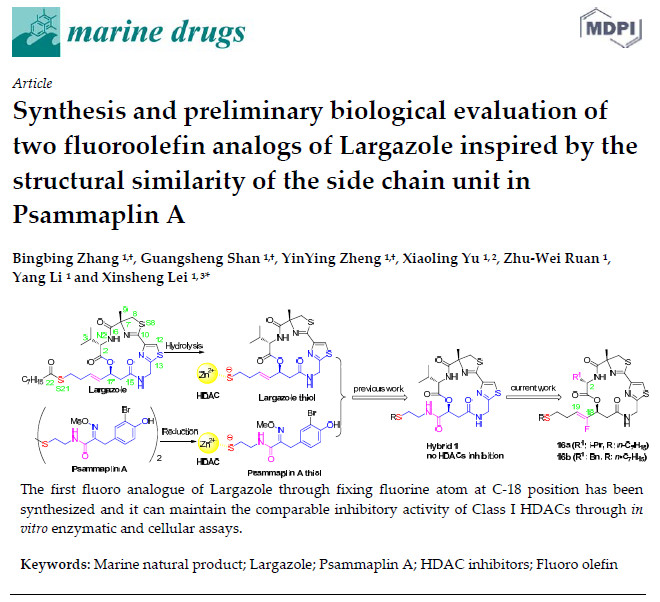
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biology
3. Materials and Methods
3.1. Chemistry
3.2. Biological Evaluation
3.2.1. Recombinant Human HDAC1, HDAC2, HDAC3, HDAC6, and HDAC8 Enzymatic Assays
3.2.2. Cytotoxic Effect Tested by 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenet. 2012, 4, 5–17. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, S.; Lauricella, M.; Tesoriere, G. Histone deacetylase inhibitors: Apoptotic effects and clinical implications. Int. J. Oncol. 2008, 33, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Verner, E.; Buggy, J.J. Isoform-specific histone deacetylase inhibitors: The next step? Cancer Lett. 2009, 280, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Tabackman, A.A.; Frankson, R.; Marsan, E.S.; Perry, K.; Cole, K.E. Structure of ‘linkerless’ hydroxamic acid inhibitor-HDAC8 complex confirms the formation of an isoform-specific subpocket. J. Struct. Biol. 2016, 195, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Taori, K.; Kim, H.; Hong, J.; Hendrik Luesch, H. Total Synthesis and Molecular Target of Largazole, a Histone Deacetylase Inhibitor. J. Am. Chem. Soc. 2008, 130, 8455–8459. [Google Scholar] [CrossRef]
- Bowers, A.; West, N.; Taunton, J.; Schreiber, S.L.; Bradner, J.E.; Williams, R.M. Thiophene-derivatized Fluorescent Benzamides as Possible Probes for Histone Deacetylases. J. Am. Chem. Soc. 2008, 130, 11219–11222. [Google Scholar] [CrossRef]
- Maolanon, A.R.; Kristensen, H.M.E.; Leman, L.J.; Ghadiri, M.R.; Olsen, C.A. Natural and synthetic macrocyclic inhibitors of the histone deacetylase enzymes. ChemBioChem 2017, 18, 5–49. [Google Scholar] [CrossRef]
- Reddy, D.N.; Ballante, F.; Chuang, T.; Pirolli, A.; Marrocco, B.; Marshall, G.R. Design and Synthesis of Simplified Largazole Analogues as Isoform-Selective Human Lysine Deacetylase Inhibitors. J. Med. Chem. 2016, 59, 1613–1633. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Tu, Z.; Liao, C.; Wang, Z.; Li, S.; Yao, H.; Li, Z.; Jiang, S. Discovery of novel class I histone deacetylase inhibitors with promising in vitro and in vivo antitumor activities. J. Med. Chem. 2015, 58, 7672–7680. [Google Scholar] [CrossRef] [PubMed]
- Clausen, D.J.; Smith, W.B.; Haines, B.E.; Wiest, O.; Bradner, J.E.; Williams, R.M. Modular synthesis and biological activity of pyridyl-based analogs of the potent Class i Histone Deacetylase Inhibitor Largazole. Bioorg. Med. Chem. 2015, 23, 5061–5074. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M.J. Action of FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum no. 968, on Ha-ras transformed NIH3T3 cells. Biosci. Biotechnol. Biochem. 1994, 58, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, N.; Ueda, H.; Takase, S.; Tanaka, H.J. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. J. Antibiot. 1994, 47, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Manda, T.; Matsumoto, S.; Mukumoto, S.; Nishigaki, F.; Kawamura, I.; Shimomura, K.J. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. III. Antitumor activities on experimental tumors in mice. J. Antibiot. 1994, 47, 315–323. [Google Scholar] [CrossRef]
- Masuoka, Y.; Nagai, A.; Shin-ya, K.; Furihata, K.; Nagai, K.; Suzuki, K.; Hayakawa, Y.; Seto, H. Spiruchostatins A and B, novel gene expression-enhancing substances produced by Pseudomonas sp. Tetrahedron Lett. 2001, 42, 411–444. [Google Scholar] [CrossRef]
- Baud, M.G.J.; Leiser, T.; Haus, P.; Samlal, S.; Wong, A.C.; Wood, R.J.; Petrucci, V.; Gunaratnam, M.; Hughes, S.M.; Buluwela, L.; et al. Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. Med. Chem. 2012, 55, 1731–1750. [Google Scholar] [CrossRef]
- Poli, G.; Fabio, R.D.; Ferrante, L.; Summa, V.; Botta, M. Largazole Analogues as Histone Deacetylase Inhibitors and Anticancer Agents: An Overview of Structure–Activity Relationships. ChemMedChem 2017, 12, 1917–1926. [Google Scholar] [CrossRef]
- Ying, Y.; Liu, Y.; Byeon, S.R.; Kim, H.; Luesch, H.; Hong, J. Synthesis and activity of largazole analogues with linker and macrocycle modification. Org. Lett. 2008, 10, 4021–4024. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yin, B.; Hu, Z.; Liao, C.; Liu, J.; Li, S.; Li, Z.; Nicklaus, M.C.; Zhou, G.; Jiang, S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org. Lett. 2010, 12, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Bowers, A.A.; West, N.; Newkirk, T.L.; Troutman-Youngman, A.E.; Schreiber, S.L.; Wiest, O.; Bradner, J.E.; Williams, R.M. Synthesis and histone deacetylase inhibitory activity of largazole analogs: Alteration of the zinc-binding domain and macrocyclic scaffold. Org. Lett. 2009, 11, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, P.; Hanigan, C.L.; Casero, R.A.; Tillekeratne, L.M. Largazole and analogues with modified metal-binding motifs targeting histone deacetylases: Synthesis and biological evaluation. J. Med. Chem. 2011, 54, 7453–7463. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Park, H.; Salvador, L.A.; Serrano, P.E.; Kwan, J.C.; Zeller, S.L.; Chen, Q.Y.; Ryu, S.; Liu, Y.; Byeon, S.; et al. Evaluation of class I HDAC isoform selectivity of largazole analogues. Bioorg. Med. Chem. Lett. 2014, 24, 3728–3731. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Ratnayake, R.; Lee, H.; Shi, G.; Zeller, S.L.; Li, C.; Luesch, H.; Hong, J. Synthesis and biological evaluation of largazole zinc-binding group analogs. Bioorg. Med. Chem. 2017, 25, 3077–3086. [Google Scholar] [CrossRef]
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, B.; Shan, G.; Wu, Y.; Yang, F.-L.; Lei, X. Synthesis of the molecular hybrid inspired by Largazole and Psammaplin A. Tetrahedron 2018, 74, 549–555. [Google Scholar] [CrossRef]
- Couve-Bonnaire, S.; Cahard, D.; Pannecoucke, X. Chiral dipeptide mimics possessing a fluoroolefin moiety: A relevant tool for conformational and medicinal studies. Org. Biomol. Chem. 2007, 5, 1151–1157. [Google Scholar] [CrossRef]
- Zoute, L.; Dutheuil, G.; Quirion, J.C.; Jubault, P.; Pannecoueke, X. Efficient Synthesis of Fluoroalkenes via Diethylzinc-Promoted Wittig Reaction. Synthesis 2006, 2006, 3409–3418. [Google Scholar] [CrossRef]
- Nagao, Y.; Hagiwara, Y.; Kumagai, T.; Ochiai, M.; Inoue, T.; Hashimoto, K.; Fujita, E. New C4-Chiral l,3-Thiazolidine-2-thiones: Excellent Chiral Auxiliaries for Highly Diastereocontrolled Aldol-Type Reactions of Acetic Acid and α,β-Unsaturated Aldehydes. J. Org. Chem. 1986, 51, 2391–2393. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, L.-P.; Jiao, X.-Z.; Liu, X.-Y.; Wu, Q.; Xie, P. Concise total synthesis of largazole. J. Asian Nat. Prod. Res. 2010, 12, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chai, H.; Su, M.-B.; Zhang, Y.-M.; Li, J.; Xie, X.; Nan, F.-J. Potent and orally efficacious bisthiazole-based histone deacetylase inhibitors. ACS Med. Chem. Lett. 2014, 5, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Luesch, H. Largazole: From discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
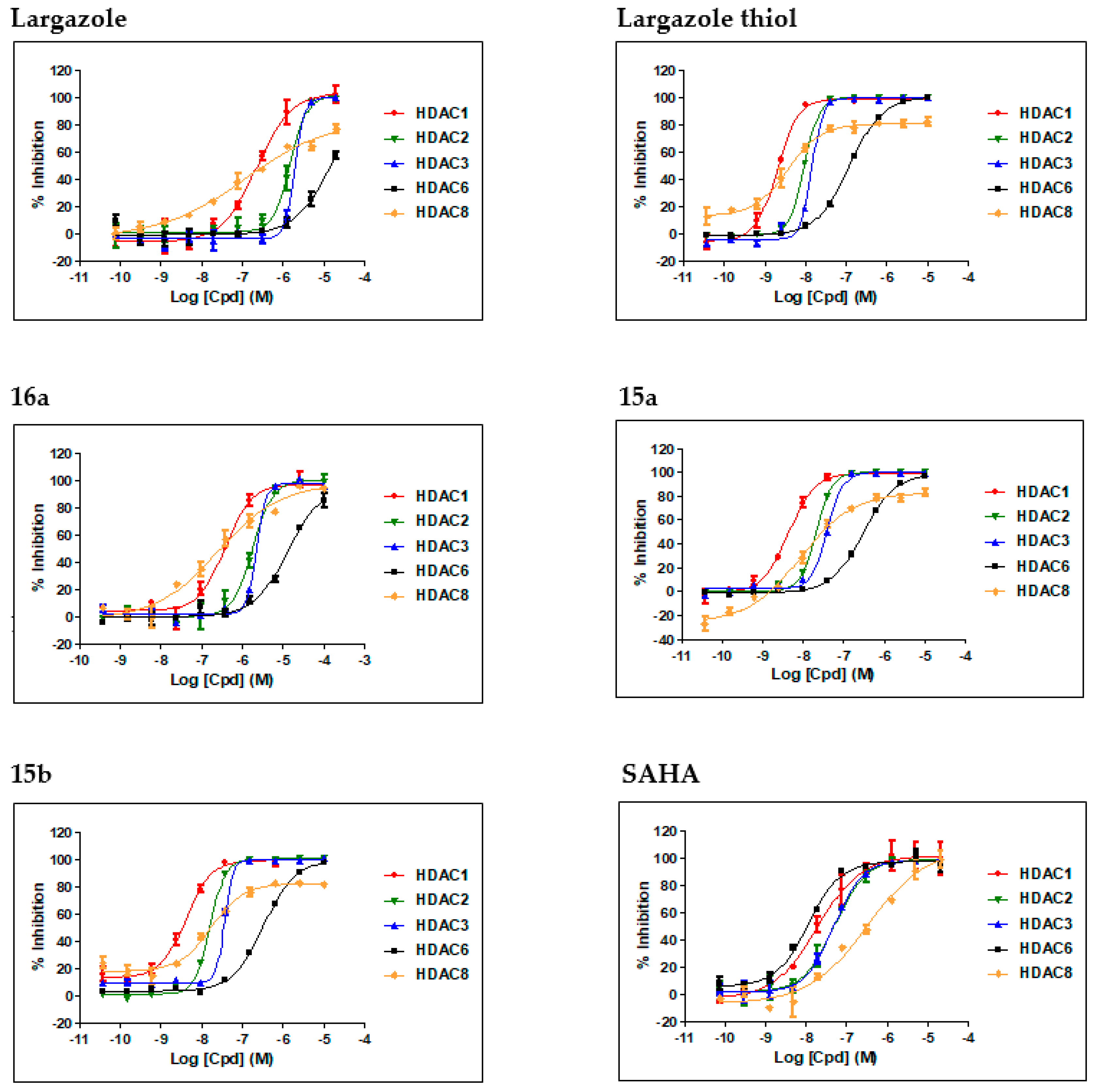
| Compound | IC50 ± SD (nM) a | Selectivity (HDAC1/6) | ||||
|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC6 | ||
| Largazole | 228 ± 34 | 1520 ± 15 | 2002 ± 28 | 120 ± 58 | >5000 | ND b |
| Largazole thiol | 2.0 ± 0.0 (1.2) c | 9.5 ± 0.2 (3.4) c | 14.4 ± 0.2 (3.5) c | 3.8 ± 0.2 (102) d | 121 ± 5.0 (49) c | 61 (40) c |
| 16a | 385 ± 45 | 1817 ± 17 | 2303 ± 25 | 255 ± 92 | >5000 | ND c |
| 15a | 4.4 ± 0.2 | 21.0 ± 0.3 | 39 ± 2 | 8.5 ± 2.5 | 300 ± 5 | 68 |
| 15b | 4.2 ± 0.3 | 16.2 ± 0.4 | 37.1 ± 0.7 | 17.6 ± 4.1 | 338 ± 17 | 80 |
| SAHA | 19 ± 4 | 51 ± 6 | 49 ± 5 | 328 ± 138 | 11 ± 1 | 0.24 |
| Compound | IC50 a ± SD (µM) | |||
|---|---|---|---|---|
| A549 | HCT116 | MDA-MB-231 | SK-OV-3 | |
| Largazole | 0.46 ± 0.10 | 0.18 ± 0.01 (0.044) b | 1.37 ± 0.41 | 0.03 ± 0.01 |
| 16a | 0.52 ± 0.12 | 0.81 ± 0.13 | 3.85 ± 0.44 | 0.25 ± 0.13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Shan, G.; Zheng, Y.; Yu, X.; Ruan, Z.-W.; Li, Y.; Lei, X. Synthesis and Preliminary Biological Evaluation of Two Fluoroolefin Analogs of Largazole Inspired by the Structural Similarity of the Side Chain Unit in Psammaplin A. Mar. Drugs 2019, 17, 333. https://doi.org/10.3390/md17060333
Zhang B, Shan G, Zheng Y, Yu X, Ruan Z-W, Li Y, Lei X. Synthesis and Preliminary Biological Evaluation of Two Fluoroolefin Analogs of Largazole Inspired by the Structural Similarity of the Side Chain Unit in Psammaplin A. Marine Drugs. 2019; 17(6):333. https://doi.org/10.3390/md17060333
Chicago/Turabian StyleZhang, Bingbing, Guangsheng Shan, Yinying Zheng, Xiaolin Yu, Zhu-Wei Ruan, Yang Li, and Xinsheng Lei. 2019. "Synthesis and Preliminary Biological Evaluation of Two Fluoroolefin Analogs of Largazole Inspired by the Structural Similarity of the Side Chain Unit in Psammaplin A" Marine Drugs 17, no. 6: 333. https://doi.org/10.3390/md17060333
APA StyleZhang, B., Shan, G., Zheng, Y., Yu, X., Ruan, Z. -W., Li, Y., & Lei, X. (2019). Synthesis and Preliminary Biological Evaluation of Two Fluoroolefin Analogs of Largazole Inspired by the Structural Similarity of the Side Chain Unit in Psammaplin A. Marine Drugs, 17(6), 333. https://doi.org/10.3390/md17060333